
| NeuroAids Vol. 3, Issue 6 (November 2000) |
The NMDA Receptor - Its Role in Neuronal Apoptosis and HIV-Associated DementiaM. Kaul1, S. A. Lipton1 1Center for Neuroscience and Aging, The Burnham Institute, 10901 North Torrey Pines Road, La Jolla, CA 92037 USA E-mail: mkaul@burnham-inst.org, slipton@burnham-inst.org . Keywords: Glutamate receptors, NMDA receptor, excitotoxicity, neurotoxicity, neuronal death, neuronal apoptosis, neurodegeneration, HIV-1-associated dementia, HIV-1, AIDS, chemokine receptors, macrophages, microglia Acknowledgments: This work was supported in part by NIH grants P01 HD29587 and R01 EY09024 (to S.A.L.) and by fellowships from the DFG and American Foundation for AIDS Research (to M.K.). We thank several present and former members of the Lipton laboratory who contributed to the work described herein, including Drs. Evan Dreyer, Peter Kaiser, Michael Yeh, Samantha Budd, Gwenn Garden, and Yun-Beom Choi, as well as members of the collaborating laboratories of Drs. Jonathan Stamler, Howard Gendelman, Pierluigi Nicotera and Sten Orrenius. |
| Abstract |
|---|
| Abstract | Introduction | Glutamate receptors, excitotoxicity and neuronal cell death | HIV-1 infection and NMDA receptor-related neuronal apoptosis | Conclusions | References | ||||
| The N-methyl-D-aspartate receptor (NMDAR) is a highly Ca2+-permeable, ligand-gated ion channel in neurons and a member of the ionotropic glutamate receptor family. Excessive stimulation of the NMDAR leads to excessive intracellular Ca2+ influx, generation of free radicals such as nitric oxide and reactive oxygen species, collapse of the mitochondrial membrane potential, loss of ATP, and eventually neuronal apoptosis or necrosis depending on the intensity of the initial insult and the extent of energy recovery. This process is termed excitotoxicity and appears to be an integral component in a final common pathway to neuronal injury in neurodegenerative disorders including HIV-associated dementia. Infection by HIV-1 is mediated by interaction of the virus' envelope protein, gp120, with chemokine receptors in addition to CD4. These HIV co-receptors are expressed on all cell types in the CNS, although microglia are the predominant if not the sole cell type that is productively infected. HIV-associated neuronal damage occurs predominantly via an indirect pathway that involves the release of various excitotoxins by macrophages. Both NMDAR antagonists and specific chemokines can protect neuron, at least in vitro, against apoptosis induced by HIV/gp120 or by NMDA, suggesting cross-talk in the signaling pathways triggered by chemokine and NMDA receptors. The present review discusses neuronal apoptosis and HIV-associated dementia in light of recent findings concerning NMDARs and chemokine receptors. |
| Introduction |
|---|
| Abstract | Introduction | Glutamate receptors, excitotoxicity and neuronal cell death | HIV-1 infection and NMDA receptor-related neuronal apoptosis | Conclusions | References | ||||
|
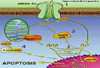
Neuronal injury and apoptosis may account, at least in part, for neurological complications associated with HIV-1 infection ranging from mild cognitive and motor impairment to dementia. The primary cell types infected in the brain are macrophages and microglia. These cells have been found in vivo and in vitro to release neurotoxic factors. Evidence has accumulated that neuronal apoptosis in HIV-related insults occurs predominantly via an indirect pathway comprising a complex cooperation of cytokines, reactive oxygen and nitrogen species, lipid mediators and excitotoxins. These molecules lead to excessive stimulation of the N-methyl-D-aspartate subtype of glutamate receptor (NMDAR). Of note, chemokine receptors, which in conjunction with CD4 mediate HIV infection of macrophages/microglia, are present on neurons and astrocytes in addition to macrophages/microglia. Thus, these receptors potentially allow direct interaction between the virus and neurons (Fig. 1).
The fact that specific chemokines ameliorate HIV/gp120-induced neuronal apoptosis that is mediated by NMDARs suggests a functional connection between the receptors for chemokines and NMDA. Accordingly, here we review the role of the NMDAR in HIV-1-related and excitotoxic neuronal cell death. |
| Glutamate receptors, excitotoxicity and neuronal cell death |
|---|
| Abstract | Introduction | Glutamate receptors, excitotoxicity and neuronal cell death | HIV-1 infection and NMDA receptor-related neuronal apoptosis | Conclusions |
|
||||
|
The NMDAR belongs to a large and heterogeneous family of membrane proteins, the glutamate receptors. These glutamate receptors recognize the major excitatory neurotransmitter in the central nervous system (CNS), (S)-glutamic acid (Glu), and other related excitatory amino acids (EAAs) (1)(2)(3). To date, four classes of EAA receptors have been identified and many member subunits cloned. These include three "ionotropic" receptor classes [iGluRs, comprised of ligand-gated ion channels termed (RS)-2-amino-3-(3-hydroxy-5-methyl-4-isoxazolyl) propionic acid (AMPA), kainic acid (KA) and NMDA receptors], and one G-protein coupled or "metabotropic" EAA receptor class [mGluRs] (1)(2)(4). Both, iGluRs and mGluRs are considered to play important roles in the CNS under normal physiologic and pathophysiologic conditions. Under physiological conditions, activation of iGluRs in neurons initiates transient depolarization and excitation. AMPARs mediate the fast component of excitatory postsynaptic currents, and NMDARs underlie a slower component. Presynaptic release of Glu and consequent depolarization of the postsynaptic neuronal membrane via AMPAR-coupled channels relieve the Mg2+ block of the ion channel associated with the NMDAR under resting conditions. This effect allows subsequent controlled Ca2+ influx through the NMDAR-coupled ion channel. This voltage-dependent modulation of the NMDAR results in activity-driven synaptic modulation (2)(5). However, extended and/or excessive NMDAR activation and consequent overexcitation can damage a neuron and eventually cause its death. This process is called excitotoxicity and appears to be favored by sustained elevation of the intracellular Ca2+ concentration and/or compromised cellular energy metabolism (5)(6). A role for Glu excitotoxicity in brain disorders was first suggested by the work of Olney following the pioneering work of Lucas and Newhouse in the retina (6). Subsequently, several lines of evidence indicated that excessive stimulation of glutamate receptors contributes to the neuropathological processes in stroke, head and spinal cord injury, Huntington's disease, Parkinson's disease, possibly Alzheimer's disease, amyotrophic lateral sclerosis, multiple sclerosis, glaucoma and HIV-1 associated dementia (1)(5)(7). Indeed, excitotoxicity seems to represent a common final pathway in a wide variety of neurodegenerative disorders (8). The NMDAR has attracted particular interest as a major player in excitotoxicity because this receptor, in contrast to most non-NMDARs (AMPA and KA receptors), is highly permeable to permeable to Ca2+, and excessive Ca2+ influx can trigger excitotoxic neuronal injury (3)(9). In addition, NMDAR antagonists effectively prevent glutamate neurotoxicity, both in vitro and in vivo in animal studies, as well as in recent phase III clinical trials with the NMDAR open-channel blocker, memantine (2)(5)(10). However, AMPA and KA receptors can also mediate excitotoxicity and contribute to neuronal damage under certain conditions (2)(5). For example, a subpopulation of Ca2+ or Zn2+-permeable AMPA receptor-coupled channels has been implicated in selective neurodegenerative disorders such as ischemia, epilepsy, Alzheimer's disease, and amyotrophic lateral sclerosis (3). Also transgenic mice overexpressing AMPARs display increased damage subsequent to ischemia when compared to control animals (11). Excessive stimulation of the NMDAR induces several detrimental intracellular signals that contribute to neuronal cell death by apoptosis or necrosis, depending on the intensity of the initial insult (12). Excessive Ca2+ influx through NMDAR-coupled ion channels leads to an elevation of the intracellular free Ca2+ concentration to a point that results in Ca2+ overload of mitochondria, depolarization of the mitochondrial membrane potential and a decrease in ATP synthesis. Additionally, excessive intracellular Ca2+ stimulates protein kinase cascades and the generation of free radicals, including reactive oxygen species (ROS) and nitric oxide (NO) (12). NO can react with ROS to form cytotoxic peroxynitrite (OONO-) (12), and in alternative redox states, NO can also activate p21ras by S-nitrosylation (transfer of the NO group to a critical cysteine thiol) (13). However, the NO group can also inhibit caspases in cerebrocortical neurons via S-nitrosylation, thereby attenuating apoptosis (16). The scaffolding protein PSD-95 (postsynaptic density-95) links the principal subunit of the NMDAR (NR1) with neuronal nitric oxide synthase (nNOS), a Ca2+-activated enzyme, and thus brings nNOS into close proximity to Ca2+ via the NMDAR-operated ion channel (14). Importantly, excessive Ca2+ influx also activates the stress-related p38 mitogen-activated protein kinase (p38 MAPK)/myocyte enhancer factor 2C (MEF2C transcription factor) pathway and c-Jun N-terminal kinase (JNK) pathways in cerebrocortical or hippocampal neurons. Activation of these pathways has been implicated in neuronal apoptosis (15)(17). As stated above, excessive intracellular Ca2+ accumulation after NMDAR stimulation leads to depolarization of the mitochondrial membrane potential (DYm) and a drop in the cellular ATP concentration. If the initial excitotoxic insult is fulminant, the cells do not recover their ATP levels and die at this point because of the loss of ionic homeostasis, resulting in acute swelling and lysis (necrosis). If the insult is more mild, ATP levels recover, and the cells enter a delayed death pathway requiring energy, known as apoptosis (12). It has been reported that NMDAR-mediated excitotoxicity leading to neuronal apoptosis also involves activation of the Ca2+/calmodulin-regulated protein phosphatase calcineurin (12), release of cytochrome c from mitochondria (18), activation of caspase-3 (19), lipid peroxidation (19), and cytoskeletal breakdown (12). Inhibition of calcineurin and caspase-3 with FK506 and caspase inhibitors, respectively, can attenuate this form of excitotoxicity (12)(19). It has been proposed that the adenine nucleotide translocator (ANT) is a part of the mitochondrial permeability transition pore (PTP) and participates in mitochondrial depolarization. Indeed, our group has found that pharmacologic blockade of the ANT with bongkrekic acid prevented collapse of the mitochondrial membrane potential (DYm), as well as subsequent caspase-3 activation and NMDA-induced neuronal apoptosis. However, treatment with bongkrekic acid failed to inhibit the transient drop in ATP concentration (although it hastened the recovery of ATP levels) and did not prevent the liberation of cytochrome c into the cytosol. Thus, initiation of caspase-3 activation and resultant neuronal apoptosis after NMDAR activation require a factor(s) in addition to cytochrome c release (18). Interestingly, stimulation of specific subtypes of the G protein-coupled mGluRs interferes with excitotoxic NMDAR-mediated activation of MAPKs and can attenuate subsequent neuronal cell death (15). Additionally, glial cells, including astrocytes, microglia and oligodendrocytes, may possess glutamate receptors (4). Both AMPA and KA receptor subtypes and mGluRs have been reported on microglia, and functional NMDARs have been reported to exist in some cases on astrocytes and oligodendrocytes. Glial glutamate receptors appear to be involved in interactions between neuronal and glial cells, and hence may conceivably contribute to synaptic efficacy. Furthermore, under certain pathologic circumstances, such as cerebral hypoxia-ischemia and possibly HIV-1 infection of the brain, astrocytes and oligodendrocytes may undergo glutamate-mediated excitotoxic cell death (4).
|
|
HIV-1
infection and NMDA receptor-related neuronal apoptosis
|
|---|
| Abstract | Introduction | Glutamate receptors, excitotoxicity and neuronal cell death | HIV-1 infection and NMDA receptor-related neuronal apoptosis | Conclusions | References | ||||
|
HIV-associated dementia eventually develops in approximately half of children and a quarter of adults infected with HIV-1 (7). Neuropathological features that may accompany this cognitive-motor complex include dendritic and synaptic damage, apoptosis and frank loss of neurons, myelin pallor, astrocytosis, and infiltration of macrophages, microglia and multinucleated giant cells (7)(20)(21). Macrophages and microglia play a crucial role in HIV-associated dementia because they are the predominant cells productively infected with HIV-1 in AIDS brains (7), although infection of astrocytes has also been rarely observed in pediatric cases (22). Also, several lines of evidence suggest that HIV-1 infected macrophages migrate into the brain (23), and the presence of macrophages/microglia has been reported to correlate with the severity of HIV-associated dementia (24). Furthermore, we and our colleagues have shown that HIV-1 infected or immune stimulated macrophages/microglia produce neurotoxins (7)(25). The mechanisms that initiate HIV-associated dementia are not completely understood. HIV-1 apparently enters the CNS soon after peripheral infection, and the virus primarily resides in microglia and macrophages, especially in those located in the perivascular space (23). It is not clear if the migration of infected monocytes and macrophages represents the only pathway for viral entry into the brain. Additionally, infection of monocytoid cells per se may not be sufficient to initiate the dementing process (23). In the CNS, HIV-1 is thought to cause immune activation of macrophages/microglia, changes in statement of cytokines, chemokines and their receptors, and up-regulation of endothelial adhesion molecules (23). However, these observations may be the result of the process rather than the inciting event for HIV-1-associated brain pathology. Therefore, it has been proposed that peripheral (non-HIV) infection or other factors may trigger events leading to dementia after HIV-1 infection in the CNS has been established. One such factor could be the increased number of activated monocytes in the circulation that express CD16 and CD69. These activated cells could possibly adhere to the normal endothelium of the brain microvasculature, transmigrate, and then trigger a number of deleterious processes (23). Infection of cells by HIV-1 can occur after binding of the viral envelope protein gp120 to one of several possible chemokine receptors in conjunction with CD4. Depending on the exact type of gp120, different HIV-1 strains may use CXCR4 (26), CCR3, CCR2, CCR5, or a combination of these chemokine receptors to enter target cells (27). Microglia are infected by HIV-1 primarily via CCR3 and CCR5 (28). CCR5 and CXCR4, among other chemokine receptors, are also present on neurons and astrocytes, and, in particular CXCR4 and CCR3 are highly expressed on neurons of macaques and humans (29). In vitro studies strongly suggest that chemokine receptors are directly involved in HIV-associated neuronal damage (17)(30)(31). Even in the absence of intact virus, the HIV proteins gp120, gp41, gp160, Tat, Nef, Rev, and Vpr have been reported to initiate neuronal damage both in vitro and in vivo (32)(33)(34)(35)(36). In this regard, the viral envelope protein gp120 has been of particular interest as it is essential for selective binding and signaling of HIV-1 to its target cell and for viral infection (17)(28)(30)(31)(34). Additionally, evidence has been provided that gp41, the membrane-spanning region of the viral envelope protein, correlates with the statement of immunologic/type II NOS (iNOS) as well as the degree of HIV-associated dementia (35). A recurring question has been whether HIV-1 or its component proteins induce neuronal damage predominantly by an indirect route, e.g., via toxins produced by infected or immune-stimulated macrophages and/or astrocytes, or by a direct route, e.g., via binding to neuronal receptors (7)(17)(37). Several lines of evidence suggest that HIV-associated neuronal injury involves predominantly an indirect route and resulting excessive activation of NMDARs with consequent excitotoxicity (25)(38)(39). Analysis of specimens from AIDS patients (39) as well as in vivo and in vitro experiments indicate that HIV-1 infection create excitotoxic conditions, most probable indirectly via induction of soluble factors in macrophage/microglia and/or astrocytes, such as glutamate-like molecules, viral proteins, cytokines, chemokines, and arachidonic acid metabolites (7)(37)(40). However, it has been suggested that HIV-1 or its protein components can directly interact with neurons and modulate NMDAR function, at least under some conditions (30)(41). Picomolar concentrations of soluble HIV/gp120 induce by in vitro and in vivo injury and apoptosis in primary rodent and human neurons (32)(34). Additionally, our group and subsequently several others have shown that gp120 contributes to NMDAR-mediated neurotoxicity (38). Both voltage-gated Ca2+ channel blockers and NMDAR antagonists can ameliorate gp120-induced neuronal cell death in vitro (38)(42). Transgenic mice expressing gp120 manifest neuropathological features that are similar to the findings in brains of AIDS patients, and in these mice neuronal damage is ameliorated by the NMDAR antagonist memantine (33)(43). It is also conceivable that other glutamate receptors in addition to NMDARs influence HIV-associated neuronal damage. In particular, disparate mGlurs have been found to up-or down-modulate excitotoxic signals triggered by NMDARs (15). In our hands, the predominant mode of neurotoxicity of HIV-1 or gp120 to cerebrocortical neurons requires the presence of macrophages/microglia; HIV-1 infected or gp120 stimulated mononuclear phagocytes have been shown to release neurotoxins that directly stimulate the NMDAR (17)(25). Those macrophage toxic factors include molecules that directly or indirectly act as NMDAR agonists, such as quinolinic acid, cysteine, platelet-activating factor (PAF), and a low-molecular weight compound named NTox (7)(40)(44). Additionally, activated macrophages/microglia and possibly astrocytes produce inflammatory cytokines, including TNF-a and IL-1b, arachidonic acid metabolites, and free radicals (ROS and NO) that may indirectly contribute to excitotoxic neuronal damage (7)(40). TNF-a and IL-1 b may amplify neurotoxin production by stimulating adjacent glial cells and by increasing iNOS activity (40)( Fig. 2).
In contrast to these indirect neurotoxic pathways, it has been reported that gp120 can directly interact with neurons in the absence of glial cells. Recently, gp120 was found to act at chemokine receptors directly on neurons to induce their death (30). Additionally, nanomolar concentrations of gp120 have been reported to interact with the glycine binding site of the NMDAR (45). Furthermore, gp120 may produce a direct excitotoxic influence via NMDAR-mediated Ca2+ oscillations in rat hippocampal neurons (46), and may bind to noradrenergic axon terminals in neocortex, where it possibly potentiates NMDA-evoked noradrenaline release (47). Nonetheless, many if not all of these direct effects on neurons were observed in vitro in the absence of glial cells. Since glial cells are known to modify these death pathways, we feel that under in vivo conditions, the indirect route to neuronal injury is the predominant one. Along these lines, gp120 has been found to aggravate excitotoxic conditions by impairing astrocyte uptake of glutamate via arachidonic acid that is released from activated macrophages/microglia (37). Metabolites of arachidonic acid, such as prostaglandins, also stimulate a Ca2+-dependent release of Glu by astrocytes (48). Moreover, HIV-1 can induce astrocytic statement of the b-chemokine known as macrophage chemotactic protein-1 (MCP-1). This b-chemokine in turn attracts additional mononuclear phagocytes and microglia to further enhance the potential for indirect neuronal injury via the release of macrophage toxins (49). HIV-1 infection and its associated neurological dysfunction involve both chemokine receptors and NMDAR-mediated excitotoxicity. This dual receptor involvement raises the question whether the G protein-coupled chemokine receptors and ionotropic glutamate receptors might influence each other's performance. Indeed, the b-chemokine known as "regulated upon activation T cell expressed and secreted" (RANTES), which binds to the chemokine receptors CCR1, CCR3 and CCR5, can abrogate neurotoxicity induced by gp120 (17) or by excessive NMDAR stimulation (50). In turn, excitotoxic stimulation can enhance expression of CCR5 (51). Whether or not these findings reflect a mechanism of feedback or crosstalk of these receptors within the brain remains to be elucidated. |
| Conclusions |
|---|
| Abstract | Introduction |
Glutamate receptors, excitotoxicity and neuronal cell death
|
HIV-1 infection and NMDA receptor-related neuronal apoptosis | Conclusions | References | ||||
|
Progress has been made in understanding the mechanisms of toxicity associated with overstimulation of NMDARs that leads to pathological neuronal excitation, excessive Ca2+ influx, and apoptosis. Increasing evidence indicates that excitotoxicity represents a common final pathway in neurological disorders, including HIV-associated dementia. NMDAR antagonists can inhibit both in vitro and in vivo the neurotoxicity of HIV/gp120 and of glutamate. Additionally, chemokine receptors, essential co-mediators of HIV infection, are present in the CNS on neurons, astrocytes and microglia, and can in part also confer protection against neuronal apoptosis induced by HIV/gp120 or NMDA. These findings suggest a functional connection between receptors for chemokines and NMDA. Recently, phase III trials with the NMDAR antagonist memantine have demonstrated benefit in a series of neurodegenerative conditions, including Alzheimer's disease and Vascular dementia. A large, multi-center clinical trial of memantine for HIV-associated dementia is currently being analyzed. In the future, clinical studies may lead to therapeutic applications of chemokines for neurodegenerative disorders as well. |
| References |
|---|
| Abstract | Introduction | Glutamate receptors, excitotoxicity and neuronal cell death |
HIV-1 infection and NMDA receptor-related neuronal apoptosis
|
Conclusions | References | ||||
|
Use the Back button in your browser to continue reading the article.
(2) Bigge CF(1999). Ionotropic glutamate receptors. Curr Opin Chem Biol. Aug; 3(4): 441-447. PMID: 10419857 Medline (3) Weiss JH, Sensi SL (2000). Ca2+-Zn2+ permeable AMPA or kainate receptors: possible key factors in selective neurodegeneration. Trends Neurosci. Aug; 23(8): 365-371. Medline (4) Gallo V, Ghiani CA (2000). Glutamate receptors in glia: new cells, new inputs and new functions. Trends Pharmacol Sci. July; 21(7): 252-258. Medline (5) Doble A (1999). The role of excitotoxicity in neurodegenerative disease: implications for therapy. Pharmacol Ther. Mar; 81(3): 163-221. Medline (6) Olney JW (1969). Brain lesions, obesity, and other disturbances in mice treated with monosodium glutamate. Science. May 9; 164(880): 719-721. Medline (7)Lipton SA, Gendelman HE (1995). Dementia associated with the acquired immunodeficiency syndrome. N Engl J Med. Apr 6; 332(14): 934-940. Medline (8) Lipton SA, Rosenberg PA (1994). Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med. Mar 3; 330(9): 613-622. Medline (9) Choi DW (1988). Glutamate neurotoxicity and diseases of the nervous. Neuron. Oct; 1(8): 623-634. Medline (10) Choi DW, Koh JY, Peters S (1988). Pharmacology of glutamate neurotoxicity in cortical cell culture: attenuation by NMDA antagonists. J Neurosci. Jan; 8(1): 185-196. PMID: 2892896 Medline (11) Le D, Das S, Wang YF, Yoshizawa T, Sasaki YF, Takasu M, Nemes A, Mendelsohn M, Dikkes P, Lipton SA, Nakanishi N (1997). Enhanced neuronal death from focal ischemia in AMPA-receptor transgenic mice. Brain Res Mol Brain Res. Dec 15; 52(2): 235-241. PMID: 9495544 Medline (12) Nicotera P, Ankarcrona M, Bonfoco E, Orrenius S, Lipton SA (1997). Neuronal necrosis and apoptosis: two distinct events induced by exposure to glutamate or oxidative stress. Adv Neurol. 72:95-101. Medline (13) Yun HY; Gonzalez-Zulueta M, Dawson VL, Dawson TM (1998). Nitric oxide mediates N-methyl-D-aspartate receptor-induced activation of p21ras. Proc Natl Acad Sci USA. May 12; 95(10): 5773-5778. Medline (14) Sattler R, Xiong Z, Lu WY, Hafner M, MacDonald JF, Tymianski M (1999). Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD-95 protein. Science. Jun 11; 284(5421): 1845-1848. Medline (15) Mukherjee PK, DeCoster MA, Campbell FZ, Davis RJ, Bazan NG (1999). Glutamate receptor signaling interplay modulates stress-sensitive mitogen-activated protein kinases and neuronal cell death. J Biol Chem. Mar 5; 274(10): 6493-6498. Medline (16)Tenneti L, D'Emilia DM, Lipton SA (1997). Suppression of neuronal apoptosis by S-nitrosylation of caspases. Neurosci Lett. Nov 7; 236(3):139-42. Medline (17) Kaul M, Lipton SA (1999). Chemokines and activated macrophages in gp120-induced neuronal apoptosis. Proc Natl Acad Sci USA. Jul 6; 96(14), 8212-8216. Medline (18) Budd SL, Tenneti L, Lishnak T, Lipton SA (2000). Mitochondrial and extramitochondrial apoptotic signaling pathways in cerebrocortical neurons. Proc Natl Acad Sci USA. May 23; 97(11): 6161-6166. Medline (19) Tenneti L, D'Emilia DM, Troy CM, Lipton SA (1998). Role of caspases in N-methyl-D-aspartate-induced apoptosis in cerebrocortical neurons. J Neurochem. Sep; 71(3): 946-959. Medline (20) Masliah E, Heaton RK, Marcotte TD, Ellis RJ, Wiley CA, Mallory M, Achim CL, McCutchan JA, Nelson JA, Atkinson JH, Grant I (1997). Dendritic injury is a pathological substrate for human immunodeficiency virus-related cognitive disorders. HNRC group. The HIV Neurobehavioral Research Center. Ann Neurol. Dec; 42(6): 963-972. Medline (21) Petito CK, Roberts B (1995). Evidence of apoptotic cell death in HIV encephalitis. Am J Pathol. May; 146(5): 1121-1130. Medline (22) Brack-Werner R, Bell JE (1999). Replication of HIV-1 in human astrocytes. Science Online: NeuroAids. Sep; 2(8). NeuroAIDS (23) Gartner S (2000). HIV infection and dementia. Science. Jan 28; 287(5453): 602-604. Medline (24) Glass JD, Fedor H, Wesselingh SL, McArthur JC( 1995). Immunocytochemical quantitation of human immunodeficiency virus in the brain: correlations with dementia. Ann Neurol. Nov; 38(5): 755-762. Medline (25) Giulian D, Vaca K, Noonan CA (1990). Secretion of neurotoxins by mononuclear phagocytes infected with HIV-1. Science. Dec 14; 250(4987): 1593-1596. Medline (26) Bleul CC, Farzan M, Choe H, Parolin C, Clark-Lewis I, Sodroski J, Springer TA (1996). The lymphocyte chemoattractant SDF-1 is a ligand for LESTR/fusin and blocks HIV-1 entry. Nature. Aug 29; 382(6594): 829-833. Medline (27) Doranz BJ, Rucker J, Yi Y, Smyth RJ, Samson M, Peiper SC, Parmentier M, Collman RG, Doms RW (1996). A dual-tropic primary HIV-1 isolate that uses fusin and the beta- chemokine receptors CKR5, CKR3, and CKR2b as fusion cofactors. Cell. Jun 28; 85(7): 1149-1158. Medline (28)He J, Chen Y, Farzan M, Choe H, Ohagen A, Gartner S, Busciglio J, Yang X, Hofmann W, Newman W, Mackay CR, Sodroski J, Gabuzda D (1997). CCR3 and CCR5 are co-receptors for HIV-1 infection of microglia. Nature. Feb 13; 385(6617): 645-649. Medline (29)Zhang L, He T, Talal A, Wang G, Frankel SS, Ho DD (1998). In vivo distribution of the human immunodeficiency virus/simian immunodeficiency virus coreceptors: CXCR4, CCR3, and CCR5. J Virol. Jun; 72(6): 5035-5045. Medline (30)Meucci O, Fatatis A, Simen AA, Bushell TJ, Gray PW, Miller RJ (1998). Chemokines regulate hippocampal neuronal signaling and gp120 neurotoxicity. Proc Natl Acad Sci USA. Nov 24; 95(24): 14500-14505. Medline (31)Meucci O, Fatatis A, Simen AA, Miller RJ (2000). Expression of CX3CR1 chemokine receptors on neurons and their role in neuronal survival. Proc Natl Acad Sci USA. Jul 5; 97(14): 8075-8080. Medline (32)Brenneman DE, Westbrook GL, Fitzgerald SP, Ennist DL, Elkins KL, Ruff MR, Pert CB (1988). Neuronal cell killing by the envelope protein of HIV and its prevention by vasoactive intestinal peptide. Nature. Oct 13; 335(6191): 639-642. Medline (33)Toggas SM, Masliah E, Rockenstein EM, Rall GF, Abraham CR, Mucke L (1994). Central nervous system damage produced by expression of the HIV--1 coat protein gp120 in transgenic mice. Nature. Jan 13; 367(6459): 188-193. Medline (34)Lannuzel A, Lledo PM, Lamghitnia HO, Vincent JD, Tardieu M (1995). HIV-1 envelope proteins gp120 and gp160 potentiate NMDA [Ca2+]i increase, alter [Ca2+]i homeostasis and induce neurotoxicity in human embryonic neurons. Eur J Neurosci. Nov 1; 7(11): 2285-93. Medline (35)Adamson DC, Wildemann B, Sasaki M, Glass JD, McArthur JC, Christov VI, Dawson TM, Dawson VL (1996). Immunologic NO synthase: elevation in severe AIDS dementia and induction by HIV-1 gp41. Science. Dec 13; 274(5294): 1917-1921. Medline (36)Nath A, Geiger JD, Mattson MP, Magnuson DS, Jones M, Berger JR (1998). Role of viral proteins in HIV-1 neuropathogenesis with emphasis on Tat. Science Online: NeuroAIDS. Oct; 1(6). NeuroAIDS (37)Lipton SA (1997). Neuropathogenesis of acquired immunodeficiency syndrome dementia. Curr Opin Neurol Jun; 10(3): 247-253. Medline (38)Lipton SA, Sucher NJ, Kaiser PK, Dreyer E B (1991). Synergistic effects of HIV coat protein and NMDA receptor-mediated neurotoxicity. Neuron July; 7(1): 111-118. Medline (39)Sardar AM, Hutson PH, Reynolds GP (1999). Deficits of NMDA receptors and glutamate uptake sites in the frontal cortex in AIDS. Neuroreport. Nov 26; 10(17): 3513-3515. Medline (40)Lipton SA (1998). Neuronal injury associated with HIV-1: approaches to treatment. Annu Rev Pharmacol Toxicol. 38: 159-77. Medline (41)Savio T, Levi G (1993). Neurotoxicity of HIV coat protein gp120, NMDA receptors, and protein kinase C: a study with rat cerebellar granule cell cultures. J Neurosci Res. Feb 15; 34(3), 265-72. Medline (42)Dreyer E B, Kaiser P K, Offermann JT, Lipton SA (1990). HIV-1 coat protein neurotoxicity prevented by calcium channel antagonists. Science. April 20; 248(4953), 364-7. Medline (43)Toggas S M, Masliah E, Mucke L (1996). Prevention of HIV-1 gp120-induced neuronal damage in the central nervous system of transgenic mice by the NMDA receptor antagonist memantine. Brain Res. Jan 15; 706(2) 303-7. Medline (44)Yeh MW, Kaul M, Zheng J, Nottet HLM, Thylin M, Gendelman H E, Lipton SA (2000). Cytokine-stimulated but not HIV-infected human monocyte-derived macrophages produce neurotoxic levels of the NMDA agonist, L-cysteine. J Immunol. April 15; 164(8), 4265-4270. PMID: 10754324 Medline (45). Fontana G, Valenti L, Raiteri M (1997). Gp120 can revert antagonism at the glycine site of NMDA receptors mediating GABA release from cultured hippocampal neurons. J Neurosci Res. Sep 15; 49(6), 732-8. Medline (46)Lo TM, Fallert CJ, Piser TM, Thayer SA (1992). HIV-1 envelope protein evokes intracellular calcium oscillations in rat hippocampal neurons. Brain Res. Oct 30; 594(2), 189-196. Medline (47)Pittaluga A, Pattarini R, Severi P, Raiteri M (1996). Human brain N-methyl-D-aspartate receptors regulating noradrenaline release are positively modulated by HIV-1 coat protein gp120. AIDS. May; 10(5), 463-8. Medline (48)Bezzi P, Carmignoto G, Pasti L, Vesce S, Rossi D, Rizzini BL, Pozzan T, Volterra A (1998). Prostaglandins stimulate calcium-dependent glutamate release in astrocytes. Nature. Jan 15; 391(6664), 281-285. Medline (49)Conant K, Garzino-Demo A, Nath A, McArthur JC, Halliday W, Power C, Gallo RC, Major EO (1998). Induction of monocyte chemoattractant protein-1 in HIV-1 tat- stimulated astrocytes and elevation in AIDS dementia. Proc Natl Acad Sci USA. Mar 17; 95(6), 3117-3121. Medline (50)Bruno V, Copani A, Besong G, Scoto G, Nicoletti F (2000). Neuroprotective activity of chemokines against N-methyl-D- aspartate or beta-amyloid-induced toxicity in culture. Eur J Pharmacol. Jul 7; 399(2-3): 117-121. Medline (51). Galasso JM., Harrison JK, Silverstein FS (1998). Excitotoxic brain injury stimulates expression of the chemokine receptor CCR5 in neonatal rats. Am J Pathol 153, 1631-1640. Medline |
| Top |
|
NeuroAids is a project of Science OnLine funded through a grant from the National Institute of Mental Health. |
|

|
Copyright ©1998 by AAAS Science Publications, Inc. |

{kind=link}