| NeuroAids Vol. 3, Issue 1 (January 2000) |
HIV-1 Infection and the Developing CNS F. Ensoli1, V. Fiorelli1 E-mail: ensolifior@axrma.uniroma1.it |
| Abstract |
|---|
| Abstract | Introduction | HIV-1
interactions with developing neural cells |
Influence
of diffusible factors |
Conclusions | References | ||||
|
Neurologic abnormalities are common in HIV-1 infected patients and may represent the dominant clinical manifestation of pediatric AIDS (1). Although neurological dysfunction has been directly related to CNS invasion by HIV-1 (2)(3), the pathogenesis of neurologic disorders remains unclear. Microglia, macrophages and astrocytes are major HIV-1 targets in the brain, whereas HIV-1 infected neurons have been rarely observed (4)(5)(6). This suggests that indirect mechanisms may account for the severe neuronal damage observed in these patients. Nevertheless, immature neuronal and glial cells, which are present during fetal development and early post natal life (7), may have different capabilities to support HIV-1 infection and replication compared to their mature counterparts. In addition, several neural trophic factors that exert critical roles in controlling neural differentiation, survival and function during embryonic development and early post-natal life (8) may play a part in regulating HIV-1 gene expression and virus replication in the developing brain. On the other hand, inflammatory cytokines and bioactive substances produced by HIV-1 infected and/or functionally activated accessory cells may concur in both regulating HIV-1 gene expression and replication (9) and directly altering neural cell survival, differentiation and function (7)(8) in the developing CNS, suggesting that HIV-1 infection during organ development may present unique features. |
| Introduction |
|---|
| Abstract | Introduction | HIV-1
interactions with developing neuronal cells |
Influence
of diffusible factors |
Conclusions | References | ||||
|
In HIV-1 infected newborn and children, central nervous system (CNS) disorders are often the result of a vertically acquired infection (1). They arecharacterized by a rapid onset of symptoms, rare occurrence of opportunistic infections and neoplasms, encephalopathy, and cortical atrophy, and are accompanied by a severe neurodevelopment retardation affecting the acquisition of both motor and cognitive milestones (1). Whether virus- or host-factors may influence the severity of the disease in newborn and infants compared to that seen in adults is, at present, unclear. The lack of evidence directly involving HIV-1 in neuronal killing both in adults and children (4)(5) suggested that functional and pathological CNS alterations, the latter including extensive demyelination and neuronal cell loss, might be due to bioactive substances produced by either HIV-1 infected or immunologically activated accessory cells such as microglia and monocyte-macrophages. Compelling evidence indicates these are major targets and a principal reservoir of the virus in both the developing and mature brain (4)(5). Nevertheless, these pathogenetic mechanisms do not explain the apparently higher incidence and severity of CNS involvement in pediatric AIDS patients compared to the adult population. One hypothesis is that the timing of HIV-1 infection may contribute to the different clinical manifestations. Presently, the proportion of infants who become infected intragestationally or are infected at the time of delivery is unclear. Throughout this period, however, the CNS is maturing and is characterized by the presence of developing neural (neuronal and glial) cells which, influenced by extrinsic regulatory signals, are differentiating toward the mature state (7)(8). Thus, the neurological impairment and retardation of neurodevelopment observed in newborns and infants, may depend upon mechanisms that, at least in part, are related to the specific requirements of actively developing and maturing cells, which may have peculiar vulnerabilities to mechanisms either directly or indirectly triggered by HIV-1. |
| HIV-1 interactions with developing neural cells |
|---|
| Abstract | Introduction | HIV-1
interactions with developing neural cells |
Influence
of diffusible factors |
Conclusions | References | ||||
|
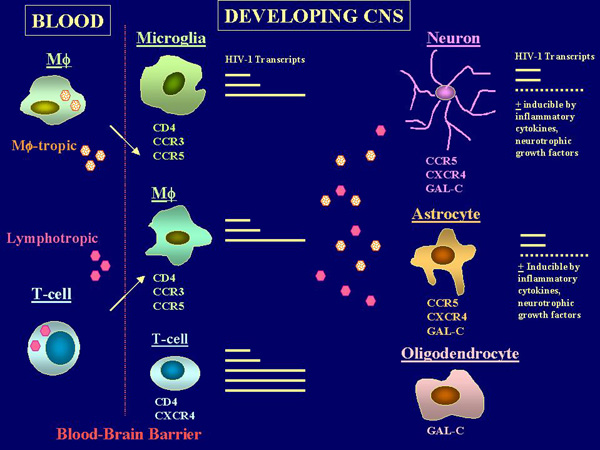
In both the developing and adult brain, HIV-1 infected neuronal and glial cells have been rarely demonstrated by conventional immunohistochemical techniques (4)(5). Recently, the use of highly sensitive techniques such as the in situ-Polymerase Chain Reaction (PCR) provided evidence for a discrete HIV-1 infection of astroglial and, to a lesser extent, neuronal cells both in adults and children, though accompanied by a low-level viral gene expression (10)(11). Indeed, the spectrum of potential HIV-1 interactions with target cells in the developing CNS appears wider than that observed in the mature brain. Pathological and in vitro studies suggest that, in addition to microglia and mononuclear phagocytes, many different neuronal and glial cell lines with an immature phenotype as well as primary neurons and astrocytes originated from the human fetal CNS appear susceptible to HIV-1 infection (12)(13)(14) through a CD4-independent pathway of virus entry. This pathway appears to involve multiple receptor components (15)(16) (Figure 1).
Interestingly, the susceptibility to HIV-1 infection and the permissivity to viral gene expression and replication in neural (neuronal and glial) cells appear to depend upon the state of cellular differentiation, and is higher with more immature precursors (14)(17) than with differentiated cells (18). The molecular features of such restricted HIV-1 infection of neuronal and glial cells as compared to their precursors have been studied in transgenic model systems (19)(20)(21)(22) as well as in vitro with cells transfected with the viral promoter/enhancer or with infectious HIV-1 proviral DNA (17)(22)(23)(24), thus bypassing restriction posed by cell binding and entry. The HIV-1 LTR includes many regulatory elements which can direct transcription in different cell types and under a variety of growth and differentiation conditions. In the CNS of transgenic mice, only the HIV-1 LTR from CNS-derived HIV-1 strains were expressed. Moreover, they were expressed almost exclusively in neurons (19)(20), suggesting that specific elements within the viral promoter can selectively and preferentially control viral gene expression in developing neuronal cells. Differentiation-dependent effects on the transcriptional activity of the HIV-1 promoter have been further characterized in the embryonic cell line NTERA-2 induced to a neuronal differentiation in vitro (23), as well as in primary fetal neuronal and glial cells or neuroblastic cell lines (17)(24)(25). These studies indicated that different patterns of utilization of regulatory elements within the viral promoter can selectively regulate viral gene expression in distinct neuronal and glial precursors, possibly through the recruitment of transcription factors that are involved in tissue-specific gene expression. This may contribute in determining preferential environments for HIV-1 gene expression in the developing CNS and could, at least in part, account for the spectrum of post-entry, cell type-associated differences in the levels of virus replication which is generally observed with neuronal and glial cells of different origin (13)(17)(26). This evidence also implies that strain-specific variation of the HIV-1 LTR may influence the ability of the virus to infect and replicate in the nervous system and may contribute in supporting viral latency or viral replication in specific cell targets within the developing brain (Figure 1). The existence of a tissue-specific evolution of the viral promoter in the CNS (27) provides further support to this hypothesis and suggests that the HIV-1 LTR may play a central role in viral tissue-specific adaptation (28)(29). It should be noted, however, that in all these cell types the levels of viral replication are generally very low (13)(17)(18), as compared to those observed with more traditional HIV-1 cell targets such as T cells or even monocyte/macrophages. Thus viral replication may be undetected by the commonly used techniques based on demonstration of viral core proteins in tissue samples as well as in the cell culture supernatants or in cytoplasmic preparations. The analysis of HIV-1 specific transcripts in infected astrocytes from fetal CNS provided some explanations to this phenomenon as it indicated that the multiply spliced 2 kb species associated with regulatory gene expression (tat, rev, and nef) represents the predominant viral mRNA species in these cells (30). Larger molecular weight mRNA associated with structural gene expression is very low and in some cases virtually absents (30). Several soluble signals, however, can modulate such permissivity and can further contribute in supporting virus latency or virus replication during organ development (9)(24)(25)(33)(34)(35). In fact, within the developing CNS, cells are under the control of environmental factors that provide instructive/permissive signals to neural cell targets (8). By regulating the survival, differentiation and maintenance of specific functions of neuronal and glial precursors, these extracellular signals can influence many steps of the CNS development and may concur in controlling virus-cell interactions in the maturing brain (25)(35). For instance, the prototypic neurotrophin nerve growth factor (NGF) can exert a positive regulation on both basal and Tat-induced LTR-directed gene expression as well as on viral replication in both immature neurons and embryonic glial cells (24). These effects of NGF appear restricted to mitotically active precursors since the addition of the neurotrophin to post-mitotic neurons did not alter viral gene expression or replication in these cells (18). These observations suggest that during embryonic and early post-natal life, differentiating cells in the nervous system can be more susceptible to HIV-1 infection and permissive to virus replication than their mature counterparts. Increased HIV-1 replication is accompanied by enhanced production of viral proteins and inflammatory cytokines, and both viral products and cytokines have been shown to induce functional and pathological alterations in both developing and mature CNS. Thus, immature neurons and glial cells may contribute in establishing a virus reservoir in the developing CNS whose extent and susceptibility to conventional antiretroviral therapy (including the more recent highly active regimens) are, at present, unknown. Whether this may participate, by either direct or indirect mechanisms, to the severity of neurological dysfunction in congenital HIV-1 infection, remains to be determined. On the other hand, the lack of measurable cytopathic effects in HIV-1 infected neuronal and glial precursors, even in the presence of relatively high levels of productive virus replication (13)(14)(17)(18), suggests that mechanism(s) other than direct virus-neural cell interactions and, possibly, additional components of the brain microenvironment are required to induce the conspicuous pathology and functional CNS damage observed in HIV-1 infected infants and children. |
| Influence of diffusible factors produced by HIV-1 infected or functionally activated accessory cells on developing neural tissues |
|---|
| Abstract | Introduction | HIV-1
interactions with developing neural cells |
Influence
of diffusible factors |
Conclusions | References | ||||
|
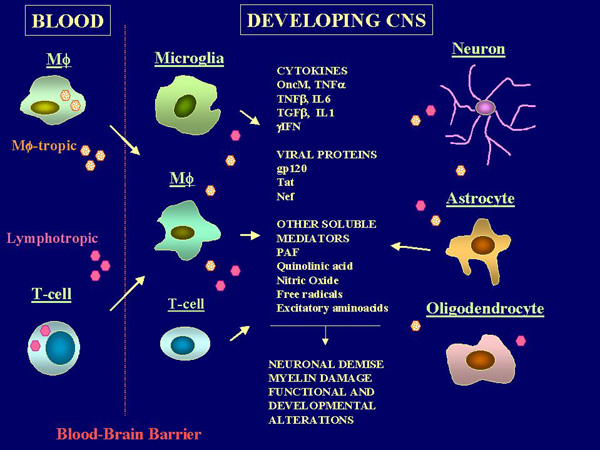
The extent of recruitment of HIV-1 infected and/or functionally activated macrophage/microglia in both the mature and developing CNS appears to better correlate to the neuropathologic changes and functional impairment than viral burden in the brain parenchyma (36)(37). Consistent with this evidence, recent studies indicated that apoptotic mechanisms of neuronal injury may be involved in HIV-1 neuropathogenesis in the absence of direct HIV-1 infection of these cells, both in vivo and in vitro (38)(39)(40). They also indicate that soluble mediators produced by peripheral blood mononuclear cells (PBMC) or enriched Monocyte/Macrophage (M/M) preparations from HIV-1 infected subjects can alter the growth and survival of immature neurons and glial cells in primary explants or dissociated cell cultures from distinct areas of the fetal CNS (41)(42)(43). Taken together, these observations suggest that the mechanism by which the virus can alter CNS development and induce pathology in the immature brain may depend upon the altered production of soluble bioactive compounds by accessory cells (Figure 2). Several potentially "neurotoxic" mediators have been identified in different model systems (31)(32)(41)(43)(44)(45)(46)(47)(48)(49)(50)(51)(52)(53). These include inflammatory cytokines, viral proteins and neurotoxic metabolites. Thus, more likely than a single "neurotoxin", a complex interaction of multiple bioactive mediators which may act in concert in altering the function and survival of actively developing and maturing cells, appears responsible for the neurologic disturbance (Figure 2).
Inflammatory cytokines Normally, the cellular expression of cytokines in the CNS is highly integrated and under a tight regulatory control which make use of the complex circuitry that represent the functional basis of the nervous system (7). Since the circuitry is designed for discrete cell to cell interactions, cytokines can regulate neural gene expression with the same degree of precision, which is inherent to the wiring. This allows for unique functional or maturational decisions by individual neurons within large groups of cell (7). However, in certain pathologic states cytokine production may become spatially and temporally dysregulated and may portend detrimental consequences (Figure 2). Altered levels of proinflammatory cytokines such as IL-1, IL-6, TNF-a and the presence of neurotoxic metabolites such as quinolinic acid (a monocyte/microglial byproduct of tryptophan with neurotoxic properties) have been documented in sera and CSF of both pediatric and adult AIDS patients (56)(57)(58). Production of the same cytokines as well as TGF-b, PAF and arachidonic acid metabolites have been also documented in the brain parenchyma, in association with HIV-1 infected and/or activated mononuclear cells and astrocytes (10)(45)(46)(47). Such soluble factors possess documented damaging potentials on both developing and mature neural tissues. In addition to mediating microglia and astrocytic activation, which in turn may lead to increased proinflammatory cytokines production (54) and virus replication (9), many of the soluble factors such as IL-1 and IL-2 can exert either a positive or a negative influence on developing oligodendrocytes by altering their proliferation and survival (59). TNF-a has been shown to mediate myelin and oligodendrocytes damage both in vivo and in vitro (60), which results in cell death. TNF-b, which is genetically and functionally related to TNF-a, has been also shown to posses potent cytotoxicity against oligodendrocytes. The effect is much more potent than TNF-a and is mediated by apoptotic mechanisms (49). IL-6 has been shown to mediate damage in developing neural tissues when expressed in the brain of transgenic mice (48). Oncostatin M (oncM), a recently characterized growth regulator which is produced by immune cells upon HIV-1 infection or functional activation, has been identified as one cytokine that can directly cause a profound inhibition of neuronal development and viability (43). Expression of oncM in transgenic mice is detrimental to CNS development, and death is associated with its expression in neuron (61), suggesting that an altered oncM production in the immature brain may alter CNS maturation and exert lethal effects on neuronal cells. Such alterations of the growth potential and survival of developing neural cells induced by different inflammatory cytokines may have important implications in our understanding the mechanism(s) responsible for the severe neurodevelopment retardation observed in HIV-1 infected infants. Indeed, such soluble mediators may exert multiple and even apparently contrasting effects upon different cell types in the brain. For example, TNFa can either exert direct inhibitory or stimulatory effects depending upon the responsive neural cell type. In fact, TNFa can even protect primary neurons of different origins from metabolic-excitotoxic insult (62). In addition, recent studies indicated that the inhibitory effects exerted by TNFa on neural cell targets may depend upon signaling interference with endogenous growth factors such as the insulin-like growth factor I (IGF-I) (63), which represents a final mediator of the biological activities controlled by the growth hormone (GH). Alterations of this pathway by inflammatory cytokines may involve additional mechanisms responsible for the growth failure and maturational defects in congenital HIV-1 infection. Viral proteins and other bioactive compounds The HIV-1 envelope glycoprotein gp120 has been shown to possess both direct and, through interactions with accessory cells, indirect excitotoxic properties in both the developing and mature brain (64)(67). In addition, gp120 can directly alter neuronal survival through interactions with the CXCR4 molecule (68), a chemokine receptor that in addition to T cells is expressed by both immature neurons and fetal astrocytes (69). The HIV-1 trans-activating protein Tat possess several neurotoxic potentials which include disturbance of signal transmission through the synaptic junction (51), direct alterations of neuronal viability (52), and altered migratory properties of immature neurons (70). The latter, in turn, depends upon mechanisms of molecular mimicry induced by the presence of RGD sequences in the 2nd exon of the tat gene. Such alterations of migratory properties may affect the spatial relationships of developing neurons and may be particularly relevant during the early phases of CNS development and maturation. In addition to directly interacting with responsive neuronal cell targets, the contribution of viral proteins to neuronal demise may involve multiple interactions with non-neuronal cell types in the brain such as microglia and astrocytes, which, upon exposure to viral proteins, can produce and release bioactive compounds with neurotoxic properties (31)(32). |
| Conclusions |
|---|
| Abstract | Introduction | HIV-1
interactions with developing neural cells |
Influence
of diffusible factors |
Conclusions | References | ||||
|
Experimental data from neural (neuronal and glial) cell lines as well as primary neural precursors suggest that the inappropriate production of cytokines and bioactive compounds that can act as instructive/permissive signals during nervous system development, or in events which involve functional or structural tissue remodeling in the brain, may have the potential to exert both direct and indirect injury in the developing CNS. However, results collected in vitro, even in primary cell culture systems, may not accurately reflect developmental and maturational events occurring in vivo, and should therefore be interpreted with caution. Nevertheless, these studies provide support to the hypothesis that a cascade of events triggered by HIV-1 infection and involving a chronic dysregulation of the expression of soluble signals is implicated in the setting of developmental and functional alterations in the immature CNS. This notion should be taken into account in future research planning, that should include relevant model systems capable of closely mimicking the developing CNS, and suggest that the development of future therapeutic strategies should be directed not only toward reducing the viral load in the brain, but also in preventing CNS invasion, controlling the deleterious effects of cytokines, and protecting neural cells from toxic metabolites. |
| Abstract | Introduction | HIV-1
interactions with developing neural cells |
Influence
of diffusible factors |
Conclusions | References | ||||
|
Use the Back button in your browser to continue reading the article. (1) Calvelli TA, Rubinstein A (1990). Pediatric HIV infection: a review. Immunodefic. Rev. 2: 83-127. Medline (2) Lyman WD, KressY, Kure K, Rashbaum WK, Rubinstein A, Soeiro R (1990). Detection of HIV in fetal central nervous system tissue. AIDS 4 (9): 917-20. Medline (3) Shaw GM, Harper ME, Hahn BH, Epstein LG, Gajdusek DC, Price RW, Navia BA, Petito CK, O'Hara CJ, Groopman JE, Cho ES, Oleske JM, Wong-Staal F, Gallo RC (1985). HTLV-III infection in brains of children and adults with AIDS encephalopathy. Science 227:177-82. Medline (4) Wiley CA, Schrier RD, Nelson JA, Lampert PW, Oldstone MBA (1986). Cellular localization of human immunodeficiency virus infection within the brains of acquired immune deficiency syndrome patients. Proc. Natl. Acad. Sci., U.S.A 83: 7089-93. Medline (5) Kure K, Llena JF, Lyman WD, Soeiro R, Weidenheim KM, Hirano A, Dickson DW (1991). Human immunodeficiency virus-1 infection of the nervous system. An autopsy study of 268 adult, pediatric and fetal brains. Human Pathol. 22: 700-10. Medline (6) Saito Y, Sharer LR, Michaels J, Mintz M, Louder M, Golding K, Cvetkovich TA, Blumberg BM (1994). Overexpression of nef as a marker for restricted HIV-1 infection of astrocytes in postmortem pediatric central nervous tissues. Neurology 44: 474-480. Medline (7) Patterson PH, Nawa H (1993). Neuronal differentiation factors/cytokines and synaptic plasticity. Cell 72: 123-37. Medline (8) Barde YA (1989). Trophic factors and neuronal survival. Neuron 2: 1525-34. Medline (9) Janabi N, Di Stefano M, Wallon C, Hery C, Chiodi F, Tardieu M (1998). Induction of human immunodeficiency virus type 1 replication in human glial cells after proinflammatory cytokines stimulation: effect of IFN-gamma, IL1-beta, and TNF-alpha on differentiation and chemokine production in glial cells. Glia 23: 304-15. Medline (10) Nuovo GJ, Gallery F, MacConnell P, Braun A (1994). In situ detection of polymerase chain reaction-amplified HIV-1 nucleic acids and tumor necrosis factor-alpha RNA in the central nervous system. Am. J. Pathol. 144: 659-66. Medline (11) Takahashi K, Wesselingh SL, Griffin DE, McArthur JC, Johnson RT, Glass JD (1996). Localization of HIV-1 in human brain using polymerase chain reaction/in situ hybridization and immunocytochemistry. Ann. Neurol. 39: 705-11. Medline (12) Harouse JM, Kunsch C, Hartle HT, Laughlin MA, Hoxie JA, Wigdahl B, Gonzalez- Scarano F (1989). CD4-independent infection of human neural cells by human immunodeficiency virus type 1. J. Virol. 63: 2527-33. Medline (13) Tornatore C, Chandra R, Berger JR, Major EO (1994). HIV-1 infection of subcortical astrocytes in the pediatric central nervous system. Neurology 44: 481-87. Medline (14) Ensoli F, Cafaro A, Fiorelli V, Vannelli B, Ensoli B, Thiele CJ (1995). HIV-1 infection of primary human neuroblasts. Virology 210: 221-25. Medline (15) Harouse JM, Bhat S, Spitalnik SL, Laughlin M, Stefano K, Silberberg DH, Gonzalez-Scarano F (1991). Inhibition of entry of HIV-1 in neural cell lines by antibodies against galactosyl ceramide. Science 253: 320-23. Medline (16) Hesselgesser J, Halks-Miller M, DelVecchio V, Peiper SC, Hoxie J, Kolson DL, Taub D, Horuk R (1997). CD4-independent association between HIV-1 gp120 and CXCR4: functional chemokine receptors are expressed in human neurons. Curr Biol. 7: 112-21. Medline (17) Ensoli F, Wang H, Fiorelli V, Zeichner SL, Luzi G, Thiele CJ (1997). HIV-1 infection and the developing Nervous System: lineage-specific regulation of viral gene expression and replication in distinct neuronal precursors. J. Neurovirology 3: 290-98. Medline (18) Sharpless N, Gilbert D, Vandercam B, Zhou JM, Verdin E, Ronnett G, Friedmann E, Dubois-Dalcq M (1992). The restricted nature of HIV-1 tropism for cultured neural cells. Virology 191: 813-25. Medline (19) Corboy JR, Buzy JM, Zink MC, Clements JE (1992). Expression directed from HIV long terminal repeats in the central nervous system of transgenic mice. Science 258: 1804-08. Medline (20) Kurth J, Buzy J.M., Lindstrom L, Clements JE (1996). In vivo transcriptional regulation of the human immunodeficiency virus in the central nervous system in transgenic mice. J Virol. 70: 7686-94. Medline (21) Buzy JM, Lindstrom LM, Zink MC, Clements JE (1995). HIV-1 in the developing CNS: developmental differences in gene expression. Virology 210: 361-71. Medline (22) Dickie P, Gazzinelli R, Chang LJ (1996). Models of HIV type 1 proviral gene expression in wild-type HIV and MLV/HIV transgenic mice. AIDS Res Hum Retroviruses 12: 1103-16. Medline (23) Zeichner SL, Hirka G, Andrews PW, Alwine JC (1992). Differentiation-dependent human immunodeficiency virus long terminal repeat regulatory elements active in human teratocarcinoma cells. J Virol. 66: 2268-73. Medline (24) Ensoli F, Ensoli B, Thiele CJ (1994). HIV-1 gene expression and replication in neuronal and glial cell lines with immature phenotype: effects of nerve growth factor. Virology 200: 668-76. Medline (25) Tornatore C, Nath A, Amemiya K, Major EO (1991). Persistent human immunodeficiency virus type 1 infection in human fetal glial cells reactivated by T-cell factor(s) or by the cytokines tumor necrosis factor alpha and interleukin-1 beta. J. Virol. 65: 6094-02. Medline (26) Dewhurst S, Sakai K, Bresser J, Stevenson M, Evinger-Hodges MJ, Volsky DJ (1987). Persistent productive infection of human glial cells by human immunodeficiency virus (HIV) and by infectious molecular clones of HIV. J. Virol. 61: 3774-82. Medline (27) Ait-Khaled M, McLaughlin JE, Johnson MA, Emery VC (1995). Distinct HIV-1 long terminal repeat quasispecies present in nervous tissues compared to that in lung, blood, and lymphoid tissues of an AIDS patient. AIDS 9: 675-83. Medline (28) Cheng-Mayer C, Weiss C, Seto D, Levy JA (1989). Isolates of human immunodeficiency virus type 1 from the brain may constitute a special group of the AIDS virus. Proc Natl Acad Sci U S A 86: 8575-9. Medline (29) McCarthy M, He J, Wood C (1998). HIV-1 strain-associated variability in infection of primary neuroglia. J. Neurovirol. 4: 80-9. Medline (30) Tornatore C, Meyers K, Atwood W, Conant K, Major EO (1994). Temporal patterns of human immunodeficiency virus type 1 transcripts in human fetal astrocytes. J Virol. 68: 93-102. Medline (31) Sawaya BE, Thatikunta P Denisova L., Brady J, Khalili K, Amini S (1998). Regulation of TNFalpha and TGFbeta-1 gene transcription by HIV-1 Tat in CNS cells. J Neuroimmunol. 87: 33-42. Medline (32) Koka P, He K, Zack JA, Kitchen S, Peacock W, Fried I, Tran T, Yashar SS, Merrill JE (1995). Human immunodeficiency virus 1 envelope proteins induce interleukin 1, tumor necrosis factor alpha, and nitric oxide in glial cultures derived from fetal, neonatal and adult human brain. J. Exp. Med. 182: 941-51. Medline (33) Vicenzi E, Poli G (1994). Regulation of HIV expression by viral genes and cytokines. J Leukoc Biol. 56: 328-34. Medline (34) Butera S (1993) Cytokine involvement in viral permissiveness and the progression of HIV disease. J Cell Biochem. 53: 336-42. Medline (35) Mizrachi Y, Rodriguez I, Sweetnam PM, Rubinstein A, Volsky DJ (1994). HIV type 1 infection of human cortical neuronal cells: enhancement by select neuronal growth factors. AIDS Res Hum Retroviruses 10: 1593-6. Medline (36) Glass J, Fedor H, Wesselingh SL, Mc Arthur JC (1995). Immunocytochemical quantification of human immunodeficiency virus in the brain: correlation with dementia. Ann. Neurol. 38: 755-62. Medline (37) Brew B, Rosenblum M, Cronin K, Price RW (1995). AIDS dementia complex and HIV-1 brain infection: clinical-virological correlations. Ann. Neurol. 38: 755-62. Medline (38) Shi B, DeGirolami U, He J, Wang S, Lorenzo A, Busciglio J, Gabuzda D (1996). Apoptosis induced by HIV-1 infection of the central nervous system. J. Clin. Invest. 98: 1979-87. Medline (39) Gelbard HA, James HJ, Sharer LR, Perry SW, Saito Y, Kazee AM, Blumberg BM, Epstein LG (1995). Apoptotic neurons in brains from paediatric patients with HIV-1 encephalitis and progressive encephalopathy. Neuropathol Appl Neurobiol. 21: 208-17. Medline (40) Petito CK, Roberts B (1995). Evidence of apoptotic cell death in HIV encephalitis. Am J Pathol. 146: 1121-30. Medline (41) Giulian D, Vaca K, Noonan CA (1990). Secretion of neurotoxins by mononuclear phagocytes infected with HIV-1. Science 250: 1593-6. Medline (42) Pulliam L, Clarke JA, McGrath MS, Moore D, McGuire D (1996). Monokine products as predictors of AIDS dementia. AIDS 10: 1495-9. Medline (43) Ensoli F, Fiorelli V, De Cristofaro M, Santini-Muratori D, Novi A, Vannelli B, Thiele CJ, Luzi G, Aiuti F (1999). Inflammatory Cytokines and HIV-1 Associated Neurodegeneration: Oncostatin-M Produced by Mononuclear Cells From HIV-1 Infected Individuals Induce Apoptosis of Primary Neurons. J. Immunol. 162: 6268-77. Medline (44) Pulliam L, Clarke JA, McGuire D, McGrath MS (1994). Investigation of HIV-infected macrophage neurotoxin production from patients with AIDS dementia. Adv Neuroimmunol. 4: 195-8. Medline (45) Wahl SM, Allen JB, McCartney-Francis N, Morganti-Kossmann MC, Kossmann T, Ellingsworth L, Mai UE, Mergenhagen SE, Orenstein JM (1991). Macrophage- and astrocyte-derived transforming growth factor beta as a mediator of central nervous system dysfunction in acquired immune deficiency syndrome. J Exp Med. 173: 981-91. Medline (46) Gelbard HA, Nottet HSLM, Swindells S, Jett M, Dzenko KA, Genis P, White R, Wang L, Choi YB, Zhang D, Lipton SA, Tourtellotte WW, Epstein LG, Gendelman HE (1994). Platelet-activating factor: a candidate human immunodeficiency virus type 1-induced neurotoxin. J. Virol. 68: 4628-35. Medline (47) Genis P, Jett M, Bernton EW, Boyle T, Gelbard HA, Dzenko K, Kene RW, Resnick L, Mizrachi Y, Volsky DJ, Epstein LG, Gendelman HE (1992). Cytokines and arachidonic metabolites produced during human immunodeficency virus (HIV)-infected macrophage-astroglia interactions: implications for the neuro pathogenesis of HIV disease. J. Exp. Med. 176: 1703-18. Medline (48) Campbell IL, Abraham CR, Masliah E, Kemper P, Inglis JD, Oldstone MBA, Mucke L (1993). Neurologic disease induced in transgenic mice by the cerebral overexpression of Interleukin-6. Proc. Natl. Acad. Sci. U.S.A. 90: 10061-5. Medline (49) Selmaj KW, Raine CS, Cannella B, Brosnam CF (1991). Identification of lymphotoxin and tumor necrosis factor in multiple sclerosis lesions. J. Clin. Invest. 87: 949-54. Medline (50) Brenneman DE, Westbrook.GL, Fitzgerald SP, Ennist DL, Elkins KL, Ruff MR, Perth CB (1988). Neuronal cell killing by the envelope protein of HIV and its prevention by vasoactive intestinal peptide. Nature 335: 639-42. Medline (51) Sabatier JM, Vives E, Mabrouk K, Benjouad A, Rochat H, Duval A, Hue B, Bahraoui E (1991). Evidence for neurotoxic activity of Tat from human immunodeficiency virus type 1. J. Virol. 65: 961-67. Medline (52) Shi B, Raina J, Lorenzo A, Busciglio J, Gabuzda D (1998). Neuronal apoptosis induced by HIV-1 Tat protein and TNF-alpha: potentiation of neurotoxicity mediated by oxidative stress and implications for HIV-1 dementia. J Neurovirol. 4: 281-90. Medline (53) Bagetta G, Corasaniti MC, Berliocchi L, Nistico R, Giammarioli AM, Malorni W, Aloe L, Finazzi-Agro A (1999). Involvement of interleukin-1beta in the mechanism of human immunodeficiency virus type 1 (HIV-1) recombinant protein gp120-induced apoptosis in the neocortex of rat. Neuroscience 89: 1051-66. Medline (54) Benveniste EN (1992). Inflammatory Cytokines within the central nervous system: sources, function and mechanism of action. Am. J. Physiol. 263: C1-C16. Medline (55) Burns TM, Clough JA, Klein RM, Wood GW, Bepman NEJ (1993). Developmental regulation of cytokine expression in the mouse brain. Growth Factors 9: 253-8. Medline (56) Mintz M, Rapaport R, Oleske JM, Connor EM, Koenigsberger MR, Denny T, Epstein LG (1989). Elevated serum levels of tumor necrosis factor are associated with progressive encephalopathy in children with AIDS. Am. J. Dis. Child. 143: 771-4. Medline (57) Hober D, Haque A, Wattre P, Beaucaire G, Mouton Y, Capron A (1989). Production of tumor necrosis factor a (TNFa) and interleukin-1 (IL-1) in patients with AIDS. Enhanced level of TNFa is related to a higher cytotoxic activity. Clin. Exp. Immunol. 78: 329-33. Medline (58) Brouwers P, Heyes MP, Moss HA, Wolters PL, Poplack DG, Markey SP, and Pizzo PA (1993). Quinolinic acid in the cerebrospinal fluid of children with symptomtic human immunodeficency virus type 1 disease: relationships to clinical status and therapeutic response. J. Inf. Dis. 168: 1380-6. Medline (59) Barres BA, Hart IK, Coles HSR, Burne JF, Voyvodic JT, Richardson WD, Raff MC (1992). Cell death and control cell survival in the oligodendrocyte lineage. Cell 70: 31-46. Medline (60) Selmaj KW, Raine CS, Farooq M, Norton WT, Brosnam CF (1991). Cytokine cytotoxicity against oligodendrocytes: apoptosis induced by lymphotoxin. J. Immunol. 147: 1522-9. Medline (61) Malik N, Haugen HS, Modrell B, Shoyab M, Clegg CH (1995). Developmental abnormalities in mice transgenic for bovine oncostatin M. Mol. Cell. Biol. 15: 2349-57. Medline (62) Cheng B, Christakos S, Mattson MP (1994). Tumor necrosis factor protects neurons against metabolic-excitotoxic insults and promotes maintenance of calcium homeostasis. Neuron 12: 139-47. Medline (63) Venters HD, Tang Q, Liu Q, VanHoy RW, Dantzer R, Kelley KW (1999). A new mechanism of neurodegeneration: a proinflammatory cytokine inhibits receptor signaling by a survival peptide. Proc. Natl. Acad. Sci. USA 96: 9879-84. Medline (64) Lipton SA (1992). Models of neuronal injury in AIDS: another role for the NMDA receptor? Trends Neurosci. 15: 75-9. Medline (65) Dawson VL and Dawson TM (1998). Nitric oxide in neurodegeneration. Prog Brain Res, 118: 215-29. Medline (66) McDonald JW, Johnston MV (1993). Excitatory amino acid neurotoxicity in the developing brain. NIDA Res. Monogr. 133: 185-205. Medline (67) Brenneman DE, Mc Cune SK, Mervis RF, Hill JM (1994). Gp 120 as an etiologic agent for NeuroAIDS: neurotoxicity and model systems. Adv. Neuroimmunol. 4: 157-65. Medline (68) Zheng J, Thylin MR, Ghorpade A, Xiong H, Persidsky Y, Cotter R, Niemann D, Che M, Zeng YC, Gelbard HA, Shepard RB, Swartz JM, Gendelman HE (1999). Intracellular CXCR4 signaling, neuronal apoptosis and neuropathogenic mechanisms of HIV-1-associated dementia. J Neuroimmunol. 98: 185-200. Medline (69) Hesselgesser J, Horuk R (1999). Chemokine and chemokine receptor expression in the central nervous system. J Neurovirol. 5: 13-26. Medline (70) Kolson DL, Buchhalter J, Collman R, Hellmig B, Farrell CF, Debouck C, Gonzalez-Scarano F (1993). HIV-1 Tat alters normal organization of neurons and astrocytes in primary rodent brain cell cultures: RGD sequence dependence. AIDS Res. Hum. Retr. 9: 677-85. Medline |
| Top |
|
NeuroAids is a project of Science OnLine funded through a grant from the National Institute of Mental Health. |
|

|
Copyright ©1998 by AAAS Science Publications, Inc. |