|
|
|
| NeuroAIDS Vol. 1, No. 3, July 1998 |
| The macaque model of HIV-induced neuroAIDS |
| O. Narayan,1 P. Cheney,2 S. V. Joag,1 E. B. Stephens,2 J. K. Marcario,2 and L. A. M. Raymond2 |
| 1Marion Merrell Dow Laboratory of Viral Pathogenesis, Department of Microbiology, Molecular Genetics, and Immunology, University of Kansas Medical Center |
| 2Mental Retardation Research Center, Department of Molecular and Integrative Physiology, Kansas City, Kansas 66160, United States |
| Address correspondence to: bnarayan@kumc.edu |
Abstract
| ||||||||||||
![]() fter briefly reviewing what is known about the four macaque SIV/SHIV viruses (SIVmac239, SIVmac251, SHIVKU-1, SHIVKU-2), the SIVmac251 model of NeuroAIDS is reviewed in detail. Specifically, the effects of this virus on the sensory system, motor output system, and behavior of infected macaques are reviewed.
fter briefly reviewing what is known about the four macaque SIV/SHIV viruses (SIVmac239, SIVmac251, SHIVKU-1, SHIVKU-2), the SIVmac251 model of NeuroAIDS is reviewed in detail. Specifically, the effects of this virus on the sensory system, motor output system, and behavior of infected macaques are reviewed.
Introduction
NeuroAIDS is the term given to the neurological complications of HIV-infection. The disease is centered around dysfunction and degeneration of neurons in the CNS that may lead to a number of clinical syndromes involving behavioral, cognitive and motor functions, abnormal evoked potentials and other electrophysiological responses.
New questions about neuropathogenesis of HIV-infection have arisen in the face of the phenomenal success that anti-HIV drugs have had in preventing AIDS. For example, does the infected CNS remain as a reservoir for the virus during suppression of virus replication in systemic tissues, i.e. does productive virus replication continue in the CNS after drugs have "shut down" systemic replication? This question is particularly relevant since many of the anti-HIV drugs do not cross the blood brain barrier. On the other hand, if productive virus replication in CNS is terminated at the same time as replication in non-neural tissues, what are the pathological residua of the inflammatory and demyelinating lesions and do neurophysiological abnormalities continue during the period of virus inhibition? Similarly, would the entire constellation of neuropathogenic effects be rekindled immediately if drug treatment is stopped? NeuroAIDS as measured by different parameters is unique to human beings since, except for chimpanzees, HIV-1 does not replicate productively in tissues of other animals. However, aspects of the disease complex have been reproduced in other animal models of lentiviral infection.
The best models now available are macaques infected either with simian immunodeficiency virus (SIV) (Letvin 1990; Letvin 1992; Letvin and King 1990; Lackner et al. 1990; Desrosiers 1990; Desrosiers and Letvin 1987; Sharma at al. 1992) or the pathogenic simian-human immunodeficiency virus (SHIV) bearing the envelope of HIV-1 (Joag et al. 1997; Joag et al. 1997b; Joag at al. 1996; Raghavan et al. 1997; Reimann et al. 1996). Similar to neurovirulent HIV-1, the defining characteristic of neurovirulent macaque viruses is their ability to replicate productively in the microglial population of the CNS. The single most dependable in vitro correlate of neurovirulence is that these viruses isolated from encephalopathic brains replicate productively in cultured macrophages from the same host species (Sharma at al. 1992; Lee and Vitetta 1992; Stephens et al. 1995; Desrosiers et al. 1991). However, as illustrated in the macaque lentivirus system, all macrophage-tropic viruses are not neurovirulent even though they may cause AIDS (Joag et al 1995; Stephens et al. 1997). This seemingly incongruous finding spawned extensive research on the genetic and phenotypic properties of the macaque viruses that cause or fail to cause neuroAIDS.
Viruses
Neurovirulent members of the lentivirus family are the only agents which replicate predominantly in microglial macrophages rather than neuroectodermal cells. Further, the lentiviral mechanisms for entry into the brain and subsequent replication in microglia are also governed by principles unique to the primate lentiviruses. Four strains of macaque viruses illustrate these unique requirements. SIVmac239 infection in rhesus macaques and SHIVKU-1 infection in pig-tailed macaques results in AIDS but not neuroAIDS (Raghavan et al. 1997; Lackner et al. 1994; Stephens et al. 1995b; Stephens et al. 1997b), whereas SIVmac251 (and similar viruses such as R71Br and 17E Br described below) and SHIVKU-2 in rhesus macaques cause AIDS and neuroAIDS (Raghavan et al. 1997; Chakrabarti et al. 1991; Lane et al. 1995; Lackner at al. 1991). All four of these viruses replicate productively in cultured, activated CD4+T cells and except for virus 239, also in cultured macrophages. SIVmac239 will infect but does not replicate productively in macrophages (Stephens et al. 1995; Mori et al. 1993). Latently or minimally productively-infected macrophages, however, can transmit the agent to lymphocytes which are permissive for replication (Zhuge at al. 1997; Zhuge at al. 1998).
Macaques inoculated with these four viruses develop infection in the CSF during the first two weeks of systemic infection. The infectivity in CSF correlates with the period of activation of infected T cells in blood (Joag at al. 1994). Even though blood also contains cell-free infectious virus in plasma at this time, only infected cells are detectable in the CSF. Among several hundred examined samples of paired blood and CSF, less than 10 CSF samples had cell-free virus (Narayan, Joag, unpublished data). Thus, infected CD4+T cells and possibly infected monocytes are the main vehicles that transmit virus from blood to the brain. Interestingly, the blood-brain-barrier is traversed without obvious infection in the vascular endothelial cells. Cultured cerebral vascular endothelial cells expressing Factor VIII are not susceptible to infection with any of the four viruses (Narayan, et al, unpublished data). Yet, all of the viruses are neuroinvasive, irrespective of whether they can replicate in macrophages. In addition to expressing viral proteins on their surfaces, these activated, infected cells express adhesion molecules that facilitate binding of the cells to vascular endothelial cells (Sasseville at al. 1992; Masih et al. 1991) and also cellular proteases such as matrix metalloproteinase-9 (Dhawan et al. 1995; Weeks et al. 1993) which (presumably) facilitate penetration of the basement membrane of the cerebral capillaries. Other virus strains (not among the four mentioned above) which are highly macrophage-lytic, do not cause activation of CD4+T cells in inoculated macaques. Such animals develop only a low grade systemic infection with minimal infection in the CSF. Brains from such animals are largely spared infection (Joag et al 1995).
The prototypic lymphocyte-tropic SIVmac239 and the dual-tropic SHIVKU-1, which do not cause neuroAIDS, invade the brain via infected CD4+T cells. The period of cellular neuroinvasion is transient and correlates with an equally transient phase of activation of CD4+T cells in blood. Resting infected T cells apparently do not cross the blood brain barrier. In SHIVKU-1-infected pig-tailed macaques, decline in the virus burden in CSF correlates with subtotal elimination of the CD4+T cell population from the animals. The transient period of pleocytosis correlates with an equally transient period of meningitis and mild perivascular cuffing in parenchymal cerebral blood vessels (Stephens et al. 1995; Lackner at al. 1991). Brains from animals infected for more than three months with either virus have no lesions and lack infectious virus in brain homogenates. However, the brains from macaques infected with either virus are positive for the presence of the viral genome by PCR (Stephens et al. 1995b). Both types of animals develop AIDS with highly productive infection and destruction in lymphoid tissues but no pathologic changes in the brain (Raghavan et al. 1997; Lackner et al. 1994; Lackner at al. 1991).
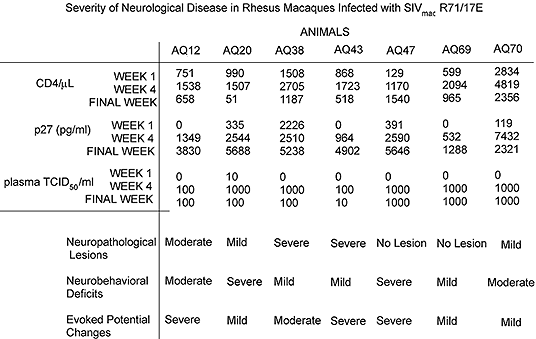
Both SIVmac251 and SHIVKU-2 cause productive infection in the CNS, the latter causing less inflammatory and more demyelinating lesions than SIVmac251 (Raghavan et al. 1997). However, more extensive studies on longitudinal infection have been performed using SIVmac251, the older of the two models. Neurovirulent SIVmac251 and the closely related SIVmac R71Br and 17EBr begin productive infection in the CNS early after inoculation of the animals and this is matched by development of extensive lentivirus-specific neuropathological changes (Chakrabarti et al. 1991; Lane et al. 1995). These in turn are matched also by abnormal physiological and behavioral findings (Table 1). These infections are acute and most animals die with AIDS and encephalitis within 16 weeks.
In summary, virus-cell interactions leading to neuroAIDS (or lack of) still remain speculative but current data suggest that: 1) In neurovirulent SIVmac251 or SHIVKU-2 infections, virus is transmitted from neuroinvading mononuclear cells to resident microglia. This event is probably mediated by binding of the viral glycoprotein expressed by the infected mononuclear cells to chemokine receptors on the microglia. 2) Virus production by microglia somehow leads to neuronal dysfunction and death. Mechanisms are speculative but include toxicity of viral gp120 for neurons, abnormal metabolism of glutamate, toxic effects of cytokines produced in brain, etc. 3) At present, no exclusive chemokine receptor has been found to be associated with neurovirulence. SIVmac239 and SIVmac251 both utilize CCR5 (Edinger at al. 1997; Chen et al. 1997) yet the two viruses are distinct with respect to neurovirulence, the former being avirulent and the latter, highly virulent. Similarly, SHIVKU-1 and SHIVKU-2 utilize CXCR4 (similar to the highly lymphocyte-tropic HIV-1 HXB2) and are highly macrophage-tropic but neither virus utilizes CCR5 (Doms, Stephens, Narayan, unpublished). Yet, the two are also distinct in neurovirulent potential, the former being avirulent and the latter, highly virulent.
SIVmac251
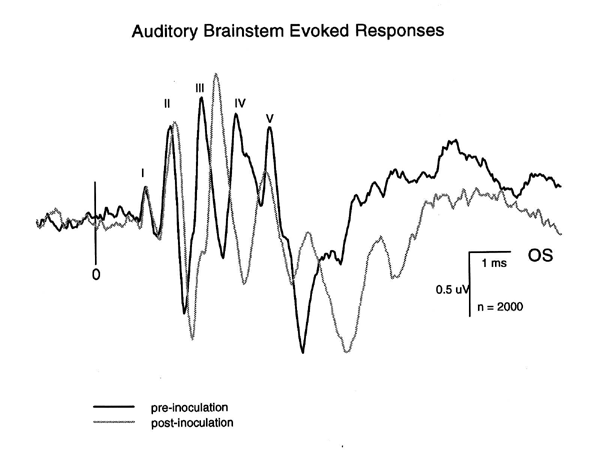
SIVmac251 and related viruses (R71Br and 17EBr) are clearly neurovirulent and cause classic lentiviral neuropathologic changes. This model has been extensively examined to determine whether demonstrable functional neurologic and behavioral deficits are associated with infection. The functional integrity sensory systems in brains of monkeys infected with neurovirulent SIVmac has been investigated by recording click-evoked auditory brainstem responses (ABR), click-evoked cortical auditory potentials (CAEP), flash-evoked visual potentials (VEP) and somatosensory-evoked potentials (SEP) from stimuli to the median nerve of the forelimb and the tibial nerve of the hindlimb (Prospero-Garcia et al. 1996; Fox et al.; Raymond et al. 1996). SIV-related abnormalities in the ABR consist primarily of conduction delays affecting preferentially the later peaks (III-V). Interpeak latency III-V shows the greatest increase suggesting more severe damage to central rather than peripheral components of the auditory pathway (Raymond et al. 1996). Figure 1 shows an example of the typical delays in ABR peak latencies associated with endstage disease in an SIV-infected monkey. Peaks II-V show increased latencies, although in this animal, only the delays in peaks III, IV and V achieved statistical significance.
| Figure 1. Superimposed ABR records from one monkey obtained during a pre-inoculation control period (10/20/95) and 8 weeks following inoculation (2/23/96). The latencies of peaks II-V are clearly increased. From Raymond et al. 1996. |
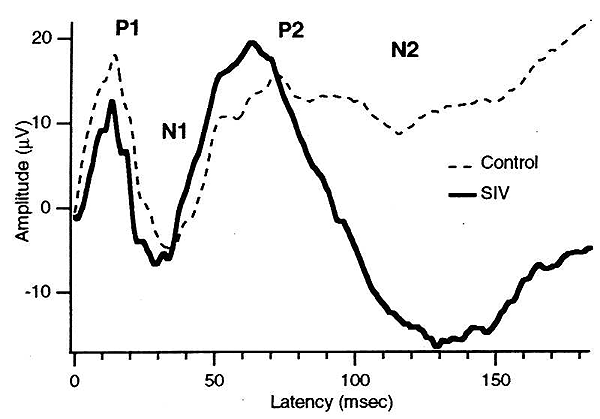
Statistically significant latency changes have been demonstrated for the ABR, CAEP and VEP using standard group statistical analysis. Peak amplitudes were not significantly changed. Single subject statistical analysis in one study (Raymond et al. 1996) showed that not all evoked potential types were abnormal in each animal. For example, from a group of seven SIV-infected animals with rapidly progressing disease, ABRs were abnormal in 4/7, VEPs in 6/7 and SEPs in 5/7. VEPs showed both latency and amplitude changes in SIV-infected animals (Prospero-Garcia et al. 1996; Fox et al.; Raymond et al. 1996). Figure 2 shows an example taken from the work of Prospero-Garcia, et al. (Prospero-Garcia et al. 1996) which illustrates shortening of the VEP P2 latency, coupled with a prominent increase in N2 latency and amplitude.
| Figure 2. (a) Average VEPs from four SIV-infected monkeys compared to the average of five control monkeys. From Prospero-Garcia et al. 1996. |
In relation to these abnormalities, it is of interest that Berman, et al. (Berman et al. 1998) found significant neuronal loss from the lateral geniculate nucleus in SIV-infected monkeys.
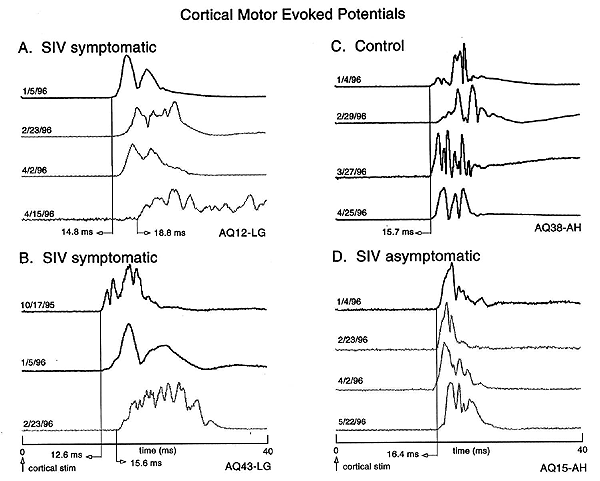
Latency increases suggestive of demyelination and conduction failure have also been demonstrated for brain motor output systems (Cheney et al. 1996). The functional integrity of the corticospinal system can be tested by recording electromyographic (EMG) responses evoked by transcranial electrical stimulation of motor cortex (so-called motor evoked potentials, MEPs). All seven of a group of SIV-infected monkeys whose disease had progressed to end stage, showed MEP onset latency increases in at least one of the four muscles tested (lateral gastrocnemius and abductor hallucis of the hindlimb and extensor digitorum communis and abductor pollicis brevis of the forelimb). Figure 3 shows examples of MEPs from four of these monkeys including an uninfected control monkey (C), two SIV-infected monkeys with rapidly progressing disease (A & B) and one SIV-infected monkey with slowly progressing disease (D).
| Figure 3. Examples of cortical MEPs recorded over a period of 4-5 months in three SIV-infected monkeys and a control monkey. Panels A and B show records for two SIV-infected monkeys with rapid progression to end stage disease. The bottom record in each panel was the final MEP obtained for that monkey and corresponded to end-stage disease. Panel C shows records for an uninoculated control monkey. Panel D shows records for an SIV-infected monkey with slowly progressing disease. This monkey remained asymptomatic over the same time period as data shown for the other monkeys. Cortical MEP latencies did not increase in either the control or asymptomatic monkeys. All monkeys were infected on 1/12/96. Light gray records were obtained after inoculation. From Cheney et al., 1996. |
Note the increases in MEP onset latency in the symptomatic monkeys with rapidly progressing disease. Similar latency increases were also found for the peripheral motor conduction pathways in MEPs evoked from stimulating the lumbar and cervical spinal cord (Cheney et al. 1996).
Table 1 gives an overall summary of the severity of evoked potential changes in monkeys with rapidly progressing SIV disease (Marcario et al. 1997). Neither of two productively infected monkeys with slowly progressing disease showed significant abnormalities in either sensory or motor evoked potentials, although one of these monkeys did develop clinical signs of systemic AIDS. All monkeys with rapidly progressing disease showed abnormal changes in sensory and motor evoked potentials. The largest latency shifts were associated with endstage disease. There was good general agreement between neuropathologic changes and the severity of evoked potential deficits, although some notable exceptions were observed.
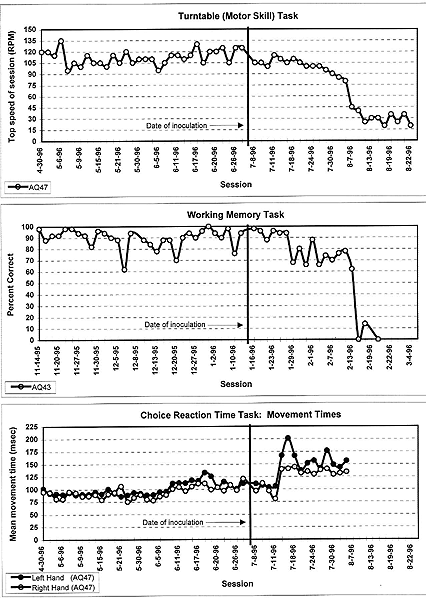
Functional impairments associated with SIV-infection in monkeys were also demonstrable using behavioral tests similar to those that have revealed deficits in HIV-1-infected humans (Fox et al.; Murray et al. 1992; Eiden et al. 1994; Marcario et al. 1996; Weed et al. 1997). Murray et al. first reported cognitive and motor deficits in a cohort of eight rhesus monkeys productively infected with SIV Delta B670. Although some monkeys showed deficits on cognitive tests of learning and memory, a motor skill task (Eiden et al. 1994) requiring the monkey to retrieve food pellets from a well in a rotating disk proved to be the most robust in revealing SIV-related impairments. Seven of eight productively infected monkeys showed deficits on this task. The motor skill task also revealed clear deficits in 5/7 monkeys infected with neurovirulent SIVmac251-like viruses (Marcario et al. 1996). Figure 4 illustrates data from one of these monkeys showing progressive decline in performance beginning about 4 weeks following inoculation.
| Figure 4. (Examples of significant impairments in performance of SIV-infected rhesus macaques one three behavioral tasks: motor skill (turntable), working memory and movement time measured from a choice reaction time task. Monkey number is given in the lower left of each panel. Time of inoculation is noted by the thick vertical line. From Marcario et al. 1996. |
Measures of sickness behavior could not account for this decline. Behavioral abnormalities were therefore attributed to SIV-related neurologic dysfunction. This conclusion is also supported by the fact that the monkey's performance on other behavioral tasks did not show deficits during this period. Fox et al. also reported deficits in a bimanual motor skill task requiring monkeys to coordinate the use of the fingers on both hands to remove raisins from a holeboard. Deficits in motor performance have also been evident from studies of reaction time and movement time using both simple and choice reaction time tasks (Marcario et al. 1996). Choice reaction time was implemented by requiring the monkey to respond with either the left or right hand depending on the color of a target "go" signal. Three of seven animals with rapidly progressing disease showed slowing in reaction time on either the simple and/or choice reaction time tasks; 6/7 animals showed significant slowing of movement time (Fig. 4), and 3/7 showed an increased error rate (e.g., exceeding the maximum allowed reaction time).
A variety of behavioral tests have been used to detect SIV-related impairments in different cognitive domains including visual recognition memory, recency memory, learning and retention (Murray et al.), spatial working memory (Fox et al.; Marcario et al. 1996), attentional set shifting (Fox et al.) and decision making time in a choice reaction time task (Marcario et al. 1996). In different SIV-infected monkeys, deficits have been detected in all of these domains suggesting a global rather than localized pathophysioloical processes in the CNS. In some cases, these deficits were slowly progressive and large (Fig. 4, working memory). However, it seems clear from all the data available, that behavioral tests focusing on cognitive function are less effective in detecting neurologic impairment than tests of motor skill (Murray et al.; Marcario et al. 1996). A possible exception to this might be measures of attentional set shifting (Fox et al.). As was true for the evoked potential data, clear disparities exist between behavioral performance deficits and the severity of neuropathologic changes (Table 1), bringing to mind similar disparities that have been noted in human beings infected with HIV-1 (Glass et al. 1993). Such disparities have also been reported by others using the SIV-infected monkey model (Fox et al.; Murray et al.; Rausch et al. 1994).
A common criticism of behavioral work in viral infected animals is whether neurologic impairments can be distinguished from performance deficits related to acute or chronic systemic disease. While this is certainly an important and difficult issue, rigorous measures of the frequency and duration of different behaviors performed by monkeys while in their home cages can provide an independent history of each monkeys "normal" behavioral profile over time (Murray et al.; Marcario et al. 1996). This can be a very informative and sensitive means of identifying the presence and onset of sickness, particularly when coupled with performance on multiple behavioral tasks and data on physiological variables such as body weight and temperature.
Conclusions
In summary, results from several studies show that sensory and motor evoked potentials and behavioral methods are effective means of detecting functional neurologic impairments in rhesus macaques infected with neurovirulent SIV and provide further validation of the SIV model of neuro-AIDS. This model may prove to be valuable for in vivo studies focusing on mechanisms of neuronal injury and development of novel drug interventions.
References
N. E. J. Berman, et al, Neuropathology and Applied Neurobiology in press, (1998).
L. Chakrabarti, et al, Am. J. Pathol. 139, 1273 (1991).
Medline
Z. Chen, P. Zhou, D. D. Ho, N. R. Landau, P. A. Marx, J. Virol. 71, 2705 (1997). Medline
P. D. Cheney, et al, Neuroscience Abstracts 22, 941 (1996).
R. C. Desrosiers, N. L. Letvin, Rev. Infect. Dis. 9, 438 (1987). Medline
R. C. Desrosiers, Annu. Rev. Immunol. 8, 557 (1990). Medline
R. C. Desrosiers, et al, Am. J. Pathol. 139, 29 (1991). Medline
S. Dhawan, et al, J. Immunol. 154, 422 (1995). Medline
A. L. Edinger, et al, Proc. Natl. Acad. Sci. U. S. A. 94, 4005 (1997). Medline
L. E. Eiden, E. A. Murray, D. M. Rausch, Neuropsychology of HIV Infection. I. Grant and A. Martin, Eds. (Oxford University Press, New York, 1994), p. 339.
H. S. Fox, L. H. Gold, S. J. Henriksen, F. E. Bloom, Neurobiol. Disease 4, 265.
J. D. Glass, S. L. Wesselingh, O. A. Selnes, J. C. McArthur, Neurology 43, 2230 (1993). Medline
S. V. Joag, et al, J. Med. Primatol. 23, 108 (1994).
S. V. Joag, et al, J. Virol. 69, 1367 (1995). Medline
S. V. Joag, et al, J. Virol. 70, 3189 (1996). Medline
S. V. Joag, et al, J. Virol. 71, 4016 (1997). Medline
S. V. Joag, et al, AIDS Res. Hum. Retroviruses 13, 635 (1997). Medline
A. A. Lackner, L. J. Lowenstine, P. A. Marx, Curr. Top. Microbiol. Immunol. 160, 77 (1990). Medline
A. A. Lackner, et al, Am. J. Pathol. 139, 609 (1991). Medline
A. A. Lackner, P. Vogel, R. A. Ramos, J. D. Kluge, M. Marthas, Am. J. Pathol. 145, 428 (1994). Medline
T. E. Lane, M. J. Buchmeier, D. D. Watry, D. B. Jakubowski, H. S. Fox, Virology 212, 458 (1995). Medline
W. T. Lee, E. S. Vitetta, J. Exp. Med. 176, 575 (1992). Medline
N. L. Letvin, N. W. King, J. AIDS 3, 1023
(1990). Medline
N. L. Letvin, Immunol. Today 11, 322 (1990). Medline
N. L. Letvin, Curr. Opin. Immunol. 4, 481 (1992). Medline
J. K. Marcario, et al, Neuroscience Abstracts 22, 941 (1996).
J. K. Marcario, et al, Neuroscience Abstracts 23, (1997).
D. T. Masih, C. E. Sotomayor, H. R. Rubinstein, C. M. Riera, Mycopathologia 114, 179 (1991). Medline
K. Mori, D. J. Ringler, R. C. Desrosiers, J. Virol. 67, 2807 (1993). Medline
E. A. Murray, D. M. Rausch, J. Lendvay, L. R. Sharer, L. E. Eiden, Science 255, 1246 (1992). Medline
O. Prospero-Garcia, et al, Proc. Natl. Acad. Sci. U. S. A. 93, 14158 (1996). Medline
R. Raghavan, et al, Brain Pathol 7, 851 (1997). Medline
D. M. Rausch, et al, J. Neuropathol. Exp. Neurol. 53, 165 (1994). Medline
L. A. M. Raymond, et al, Neuroscience Abstracts 22, 941 (1996).
K. A. Reimann, et al, J. Virol. 70, 6922
(1996). Medline
V. G. Sasseville, et al, Am. J. Pathol. 141, 1021 (1992). Medline
D. P. Sharma, et al, J. Virol. 66, 3550 (1992). Medline
E. B. Stephens, H. M. McClure, O. Narayan, Virology 206, 535 (1995). Medline
E. B. Stephens, et al, J. Leukoc. Biol. 62, 12 (1997). Medline
E. B. Stephens, et al, Virology 213, 600 (1995). Medline
E. B. Stephens, et al, Virology 231, 313 (1997). Medline
M. R. Weed, et al, Neuroscience Abstracts 23, (1997).
B. S. Weeks, et al, AIDS Res. Hum. Retroviruses 9, 513 (1993). Medline
W. Zhuge, F. Jia, I. Adany, O. Narayan, E. Stephens, Virology 227, 24 (1997). Medline
W. Zhuge, et al, Submitted (1998).
Wiley CA, Soontornniyomkij V, Radhakrishnan
L, Masliah E, Mellors J, Hermann SA, Dailey P, Achim CL (1998) Distribution
of brain HIV load in AIDS. Brain Pathology 8(2): 277-284
Medline
| Copyright Information | Site map |
{kind=link}