
VIROLOGY:
Weapons of Mutational Destruction
Vineet N. KewalRamani and John M. Coffin*
The remarkable array of defenses that cells use to fight viral infections is rapidly becoming more visible. An oft-proposed strategy for fighting viral infections is to design drugs that induce a high rate of mutation, potentially causing the viruses to succumb to "error catastrophe." A recent cluster of papers (1-5) describes a new function for a cellular protein called APOBEC3G that may act in just this way to block the replication of retroviruses such as human immunodeficiency virus (HIV).
Researchers have long recognized that the viral protein Vif is essential for replication of HIV in "nonpermissive" primary human CD4+ T cells, as well as in some transformed T cell lines (6-8). But it remained unclear how Vif enables HIV to replicate in nonpermissive cells. Vif appeared to exert its effect in either the production or transmission of new virus particles (9-11). Although virus particles produced by nonpermissive cells in the absence of Vif appear to be physically indistinguishable from those produced in its presence, their ability to infect any host cell type is greatly diminished (12-14). Initial examination of the replication block indicated that HIV produced in nonpermissive cells without Vif could not efficiently complete reverse transcription of the viral RNA genome (10, 11, 15). Elegant experiments have shown that the nonpermissiveness of cells is a dominant effect and that Vif's ability to counteract it is species specific (16-18). These results and others implied the presence of a dominant factor in nonpermissive cells that reduces virus infectivity and that is counteracted by Vif.
In 2002, Sheehy and colleagues (19) identified this inhibitory protein as CEM15, later called APOBEC3G. This protein belongs to a family of nucleic acid editing enzymes related to APOBEC1, a cytidine deaminase that edits the apolipoprotein B messenger RNA (mRNA). Expression of APOBEC3G, normally restricted to nonpermissive cell types, converts permissive cells into nonpermissive cells. The APOBEC enzyme family deaminates specific cytidine (C) residues in either DNA or mRNA, converting them to uridine (U) residues (19, 20). In addition to APOBEC1, the family includes the activation-induced cytidine deaminase (AID), a protein involved in the generation of antibody diversity. Expression of either APOBEC1 or APOBEC3G in Escherichia coli greatly increases the rate of C T mutations in the DNA (20). This observation suggested that deamination of cytidines in either the HIV RNA genome or its DNA copy might mediate the antiviral effect of APOBEC3G. Such a mechanism would require that APOBEC3G targets retroviral particles and that Vif regulates this process. Both hypotheses have been borne out by a series of recent papers that appeared rapidly after the identification of APOBEC3G as the cellular target of Vif (1-5). T mutations in the DNA (20). This observation suggested that deamination of cytidines in either the HIV RNA genome or its DNA copy might mediate the antiviral effect of APOBEC3G. Such a mechanism would require that APOBEC3G targets retroviral particles and that Vif regulates this process. Both hypotheses have been borne out by a series of recent papers that appeared rapidly after the identification of APOBEC3G as the cellular target of Vif (1-5).
In the first report, published in Science, Lecossier et al. (2) found a marked increase in guanine (G) to adenine (A) mutations in newly synthesized HIV DNA following infection with Vif-defective HIV produced by nonpermissive cells. Despite the inferred presence of APOBEC3G in virions, no effect on HIV genomic RNA was observed. These results have been reproduced and extended through direct examination of APOBEC3G activity (1, 3-5). Coexpression of APOBEC3G during Vif-defective HIV production in permissive cells strongly impaired infectivity of viral progeny. However, coexpression of a catalytically inactive APOBEC3G mutant protein had no effect on the infectivity of Vif-defective HIV (3, 5). As noted by Sheehy et al. (19), APOBEC3G copurifies in HIV virions produced in the absence of Vif (1, 3, 4). Although APOBEC3G appears to be largely confined to the cell cytoplasm (3), it is not clear how APOBEC3G becomes incorporated into HIV particles. Remarkably, APOBEC3G can be incorporated into the virions of unrelated retroviruses, including murine leukemia virus (MLV) (1, 3, 4), and its antiviral effect can be counteracted by the Vif of HIV-1 (1, 3, 17).
All of these results support a model (see the figure) wherein APOBEC3G is incorporated into virions by nonspecific interactions (with the RNA genome, perhaps) and then deaminates cytidine residues specifically on the minus DNA strand (synthesized by reverse transcribing the RNA genome). When copied into plus-strand DNA, the mutations appear as the complementary GA changes. The apparent specificity for minus-strand targets could reflect APOBEC3G's preference for single-stranded DNA or DNA-RNA hybrids. However, it is more likely that deoxyuridine is also introduced into plus-strand DNA when it is double-stranded. Unlike single-stranded DNA or an RNA-DNA hybrid minus strand, double-stranded DNA is rapidly repaired by the cellular repair machinery.
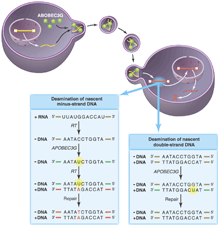
APOBEC3G on the warpath. Retrovirus production in a nonpermissive cell expressing APOBEC3G results in the incorporation of APOBEC3G (green) into viral progeny. (Left box) Although these virions are able to invade host cells and undergo the early steps of reverse transcription, the APOBEC3G cytidine deaminase induces rapid accumulation of deoxyuridine residues (green) at mutagenic hotspots in the nascent minus strand of the transcribed retroviral DNA. These mutations diminish second-strand DNA synthesis by reverse transcriptase, increasing the lability of the viral genome. Copying of the mutated base into plus-strand DNA and repair by cellular systems makes the mutation permanent (red) and integration of the viral DNA genome proceeds, resulting in a hypermutant provirus. (Right box) Mutations introduced into double-stranded DNA are likely to be repaired correctly. The presence of the viral protein Vif in the infected host cell prevents the incorporation of APOBEC3G into retroviral particles.
CREDIT: KATHARINE SUTLFIFF/SCIENCE
The distribution of mutations introduced in this way reveals an interesting preference (1-5, 20) for runs of at least two Gs, most often preceded by a T or A residue, corresponding to deamination in runs of two or more Cs followed by an A or T residue. As an antiviral mechanism this preference might be particularly efficient because mutation of either or both Gs in a TGG (tryptophan) codon leads to a translation termination signal. Thus, nearly one out of three such mutations would be lethal to the virus. Another lethal event could result from the negative effects of uridine residues in viral DNA on reverse transcription (1, 3-5, 15, 21). Hypermutant viral genomes and proviruses with mutant spectra similar to those observed in these studies have been repeatedly identified both in virus-infected cell cultures and in HIV patients (22-27). Initial suggestions that these hypermutant viral genomes resulted from errors introduced by reverse transcriptase or an imbalance in the host nucleotide pool clearly need revision.
Thus, APOBEC3G seems to be an innate viral restriction mechanism. Attacking viral genomes is a tried and true host defense: Long ago, bacteria evolved restriction endonucleases to cleave the DNA of the phage that infected them. Similarly, vertebrate cells shut off cellular protein synthesis when they detect double-stranded (viral) RNA (28). Many organisms use small interfering RNAs (siRNAs) as an antiviral defense. These defense mechanisms make a direct assault within the newly infected cell, whereas APOBEC3G is a letter bomb slipped into the virus particle on its way out of the host cell.
Unlike other family members, APOBEC3G has not been ascribed a normal cellular function, raising the possibility that it evolved specifically as an antiviral (perhaps even antiretroviral) defense. In support of APOBEC3G's involvement in innate immunity, Mariani and colleagues recently identified and characterized the murine, macaque, and African green monkey orthologs of this deaminase and found that they all efficiently restricted HIV replication (4). The inability of HIV-1 Vif to counteract any but human APOBEC3G may provide a significant species-specific barrier to primate lentivirus transmission.
We have much to learn about how Vif counteracts APOBEC3G action. It remains unclear how APOBEC3G targets retrovirus particles, and, reciprocally, how Vif excludes APOBEC3G from HIV virions. Mariani et al. (4) demonstrate that Vif interacts directly with APOBEC3G; this interaction could lead to sequestration, inactivation, or degradation of the target protein, or its exclusion from the virion. In fact, APOBEC3G degradation products have been detected in the presence of Vif, but Vif does not appear to affect the half-life of APOBEC3G within cells (4).
Does APOBEC3G's ability to block HIV infectivity extend to other retroviruses? Recent studies by two groups suggest that overexpression of human APOBEC3G during production of MLV vectors resulted in its incorporation into viral particles. This resulted in GA hypermutation in newly synthesized MLV DNA and impaired viral replication (1, 3). Significantly, Vif appeared to counteract the deleterious effect of human APOBEC3G on MLV replication. In a third study, human APOBEC3G was efficiently incorporated into replication-competent MLV particles, but there was a less dramatic effect on viral replication (4). In addition, MLV replication was not blocked by murine APOBEC3G, suggesting that simple retroviruses have developed as yet unidentified measures to counter cytidine deaminases.
Although human APOBEC3G is expressed in primary immune lymphocytes (19), its full spectrum of expression has not been finely examined. It is also unclear whether APOBEC3G expression is up-regulated during viral infection and whether it can be activated artificially. It will be interesting to discover whether there are individuals expressing unusually low or high levels of APOBEC3G or related cytidine deaminases and whether they are more or less susceptible to HIV infection. It has been noted that there are GA hypermutations in simple retroviruses, HIV-1 and -2, and hepadnaviruses (22-26, 29). Thus, it will be intriguing to determine whether APOBEC3G has contributed to the genetic diversity of retroviruses, and to test whether cytidine deaminases are broadly lethal against other virus genera. Clearly, the search for antiviral weapons is far from over. Interested parties are encouraged to stay tuned, or, better yet, to join in the hunt.
References
- R. S. Harris et al., Cell 113, 803 (2003) [Medline].
- D. Lecossier et al., Science 300, 1112 (2003).
- B. Mangeat et al., Nature 424, 99 (2003) [Medline].
- R. Mariani et al., Cell 114, 21 (2003) [Medline].
- H. Zhang et al., Nature 424, 94 (2003) [Medline].
- M. Emerman, M. H. Malim, Science 280, 1880 (1998).
- K. Strebel et al., Nature 328, 728 (1987) [Medline].
- A. G. Fisher et al., Science 237, 888 (1987) [Medline].
- D. H. Gabuzda et al., J. Virol. 66, 6489 (1992) [Medline].
- J. H. Simon, M. H. Malim, J. Virol. 70, 5297 (1996) [Medline].
- U. von Schwedler et al., J. Virol. 67, 4945 (1993) [Medline].
- N. C. Gaddis et al., J. Virol. 77, 5810 (2003) [Medline].
- R. A. Fouchier et al., J. Virol. 70, 8263 (1996) [Medline].
- C. Ochsenbauer et al., J. Gen. Virol. 78, 627 (1997) [Medline].
- J. Goncalves et al., J. Virol. 70, 8701 (1996) [Medline].
- J. H. Simon et al., J. Virol. 69, 4166 (1995) [Medline].
- J. H. Simon et al., EMBO J. 17, 1259 (1998) [Medline].
- N. Madani, D. Kabat, J. Virol. 72, 10251 (1998) [Medline].
- A. M. Sheehy et al., Nature 418, 646 (2002) [Medline].
- R. S. Harris et al., Mol. Cell 10, 1247 (2002) [Medline].
- G. J. Klarmann et al., J. Biol. Chem. 278, 7902 (2003) [Medline].
- F. Gao et al., Nature 358, 495 (1992) [Medline].
- Y. Li et al., J. Virol. 65, 3973 (1991) [Medline].
- V. K. Pathak, H. M. Temin, Proc. Natl. Acad. Sci. U.S.A. 87, 6019 (1990) [Medline].
- J. P. Vartanian et al., J. Gen. Virol. 83, 801 (2002) [Medline].
- J. P. Vartanian et al., J. Virol. 65, 1779 (1991) [Medline].
- S. Wain-Hobson et al., Virology 209, 297 (1995) [Medline].
- M. J. Korth, M. G. Katze, Curr. Top. Microbiol. Immunol. 242, 197 (2000) [Medline].
- S. Gunther et al., Virology 235, 104 (1997) [Medline].
V. N. KewalRamani is in the HIV Drug Resistance Program, National Cancer Institute, Frederick, MD 21702, USA. E-mail: vineet@ncifcrf.gov J. M. Coffin is in the Department of Molecular Biology and Microbiology, Tufts University, Boston, MA 02111, USA. E-mail: john.coffin@tufts.edu
10.1126/science.1088965
Include this information when citing this paper.
Volume 301,
Number 5635,
Issue of 15 Aug 2003,
pp. 923-925.
Copyright © 2003 by The American Association for the Advancement of Science. All rights reserved.
|