|
|
|
| AIDScience Vol. 1, No. 17, December 2001 |
| HIV-1 traffic: cell-free, cell-associated and other CNS activating stories |
| By Yuri Persidsky and Anuja Ghorpade |
| The Center for Neurovirology and Neurodegenerative Disorders, Department of Pathology and Microbiology, 985215 Nebraska Medical Center, Omaha, Nebraska 68198-5215, United States. |
| Address correspondence to: ypersids@unmc.edu or aghorpad@unmc.edu |
Abstract
| ||||||||
![]() ssues of HIV-1 trafficking into the central nervous system (CNS) are closely linked with entry of cells from the periphery into the CNS, their immune status, and the resultant immune activation of neural cells. In addition, molecular evolution of HIV-1 in different tissue compartments and the changes of acquired immune responses to virus influence these phenomena. Although the mode of HIV-1 penetration of the CNS may be diverse, there is general consensus that monocyte-associated virus is the predominant route of HIV-1 traffic in the brain. Mononuclear phagocytes (MPs) deliver the virus to the brain acting as a "Trojan horse." Neuro-immune events in late stage HIV-1-associated dementia (HAD) mainly revolve around MP secretory products (cellular and viral toxins) produced during HIV-1 encephalitis. Thus, indirect mechanisms initiated by activated MPs play a major role in neuronal dysfunction, resulting in HAD.
ssues of HIV-1 trafficking into the central nervous system (CNS) are closely linked with entry of cells from the periphery into the CNS, their immune status, and the resultant immune activation of neural cells. In addition, molecular evolution of HIV-1 in different tissue compartments and the changes of acquired immune responses to virus influence these phenomena. Although the mode of HIV-1 penetration of the CNS may be diverse, there is general consensus that monocyte-associated virus is the predominant route of HIV-1 traffic in the brain. Mononuclear phagocytes (MPs) deliver the virus to the brain acting as a "Trojan horse." Neuro-immune events in late stage HIV-1-associated dementia (HAD) mainly revolve around MP secretory products (cellular and viral toxins) produced during HIV-1 encephalitis. Thus, indirect mechanisms initiated by activated MPs play a major role in neuronal dysfunction, resulting in HAD.
Introduction
CNS disease is a common complication of late stage HIV-1 infection. Before the era of highly active antiretroviral therapy (HAART), about half of virus-infected individuals developed neuropathological changes, discovered during autopsy, and a quarter had a triad of clinical cognitive, behavioral and motor abnormalities ranging from mild motor/cognitive deficits to overt dementia (HIV-1 associated dementia [HAD]) (1, 2). Despite the diminished incidence of HAD, currently effecting 11% of those with HIV-1 (3), the greater life expectancy of infected individuals suggests that the prevalence of such complications of viral infection will continue unabated as a consequence of viral mutation and/or failure or intolerance of HAART (4). A proportional increase in HAD compared with other AIDS-defining illnesses and a marked increase in the median CD4 cell count at HAD diagnosis have occurred since the introduction of HAART (5). Despite the high incidence of neurological disease, the host-viral interactions leading to HAD are not well understood. While HAD is associated with productive viral infection of brain mononuclear phagocytes (MPs; perivascular and parenchymal macrophages and microglia) (6), there is limited evidence for infection in macroglia (astrocytes and oligodendrocytes) or in neurons (7, 8). Clinical disease is often, but not always, correlated with neuropathologic features of HIV-1 encephalitis (HIVE) (productive infection of brain macrophages and microglia, giant cell formation, and macrophage infiltration into the brain) and neocortical atrophy (neuronal loss, dendritic arbor damage, and spatial neuron alterations) (9-11). Glass et al. (12), demonstrated that the best histopathologic correlate of HAD is the number of inflammatory MPs in the CNS. Interestingly, most patients with HIVE have HAD, but not all HAD patients have HIVE (12). Even minimal increases in the numbers and, perhaps even more importantly, the state of MP immune activation could be sufficient to cause neurological dysfunction. Activated MPs produce a variety of neurotoxins including arachidonic acid and its metabolites, platelet-activating factor, pro-inflammatory cytokines (TNF- or IL-1
or IL-1 ), quinolinic acid, NTox, and nitric oxide (13-17). Viral proteins such as gp120 (18), gp41 (17), and Tat (19), or virion binding to chemoattractive cytokine (chemokine) receptors expressed on neurons can also affect neuronal viability and/or function (20, 21). Indeed, there is widespread microglial activation and accompanying reactive astrogliosis in the areas with pronounced dendritic damage (22, 23). Thus, the initial events that lead to entry of the virus into the brain culminate in disease progression and MPs likely play an important role in both these processes.
), quinolinic acid, NTox, and nitric oxide (13-17). Viral proteins such as gp120 (18), gp41 (17), and Tat (19), or virion binding to chemoattractive cytokine (chemokine) receptors expressed on neurons can also affect neuronal viability and/or function (20, 21). Indeed, there is widespread microglial activation and accompanying reactive astrogliosis in the areas with pronounced dendritic damage (22, 23). Thus, the initial events that lead to entry of the virus into the brain culminate in disease progression and MPs likely play an important role in both these processes.
HIV-1 neurotropism and cell-free viral traffic
Progressive HIV-1 infection of the human brain involves clinical sequelae including neurological and psychiatric abnormalities (4). Thus, from the viewpoint of CNS pathogenesis, HIV-1 is neurotropic in ways similar to other lentiviruses that cause diseases of the CNS (24). However, a virology-based definition would necessitate infection of neurons to fulfill the criteria of neurotropism. This does not occur in HIV-1 infection of the CNS, a fact generally accepted by the scientific community. In our minds, although this is simply a matter of semantics, HIV-1 would be better described as being neurovirulent or neuroinvasive rather than neurotropic (25). This is further supported by the notion that all in vitro definitions of neurotropic HIV-1 are based on macrophage-tropism assays of brain-derived isolates.
Earlier work that compared the molecular evolution of neuroinvasive HIV-1 sequences showed that matched comparisons of brain-derived isolates with those from different tissues exhibit differences in env, tat and pol genes (26-31). This led to the belief that HIV-1 enters the CNS soon after infection, is sequestered in CNS tissue, and undergoes a compartmentalized molecular evolution. These hypotheses were supported by data showing that brain-derived isolates from different individuals were evolutionarily closer than brain- and blood-derived isolates within an individual (32-34). In addition to macrophage-monocyte tropism, a signature brain-derived pattern was observed in isolates obtained from stereotactic brain biopsy specimens of patients. HIV-1 env sequences especially exhibit higher frequency of mutations. A variety of reasons, including viral recombination, have been utilized to explain this observation (30). The majority of these early studies were performed with clade B of HIV-1 isolates. The degree of diversity in clade B blood-derived HIV-1 isolates was shown to correlate with the severity of disease progression (35); further investigations of other clades of HIV-1 such as clade A and D were performed. The premise for these studies was based on the notion that clade A and D isolates from Asia, Africa, and other parts of the world demonstrate a more rapid disease progression and shorter time intervals from the point of seroconversion to AIDS. The data from HIV-1 isolates from sub-Saharan Africa and southeast Asia demonstrate that sequence diversity in clade A and D are dependent on specific domains of the viral genome (36). The V3 and V4 regions of gp120 had greater diversity in the brain and spleen among clade D viruses as compared to clade A; the V1 region of gp120 had the reverse phenomenon. The data indicate that selective pressures may operate differentially on various viral sequence domains. Interestingly, in multiple studies with molecularly diverse isolates of HIV-1, a significant preference for use of CCR5 as a co-receptor was observed. In addition, the V3 region of clade B X4-like sequences is more variable than the comparable clade B R5-like HIV-1 sequences (37). An overwhelming amount of literature has emerged since the discovery that HIV-1 uses chemokine receptors in conjunction with CD4 for infection (36, 38-46). Consistent with many of these reports, several brain quasispecies of HIV-1 demonstrate a preference for use of CCR5, although CCR5 utilization is not the signature of a neurovirulent isolate (47).
Monocyte-associated HIV-1 traffic
The most widely accepted mechanism by which HIV-1 enters the brain is penetration of virus-infected monocytes or CD4+ lymphocytes across the blood-brain barrier (BBB). Several mechanisms were suggested as potentially important in this regard, including increased production of pro-inflammatory substances, cytokines, or viral proteins in peripheral blood of HIV-1 infected patients (48, 49), resulting in immune activation of brain microvascular endothelial cells (BMVEC). This leads to the up-regulation of adhesion molecules on BMVEC promoting MP CNS migration (50). Alternatively, peripheral immune activation could influence the selection of specific subsets of monocytes, which have selective advantage to traverse the BBB (51). Indeed, immune activated MPs migrated in larger numbers through artificial BBB models than non-activated cells (52). Successful penetration of the BBB requires expression of metalloproteinases (MMPs), proteolytic enzymes responsible for the integrity and turnover of the extracellular matrix. Increased concentrations of MMP-9 were found in cerebrospinal fluid (CSF) of HIV-1 infected patients with neurological impairment, as compared to patients without it (53). Analysis of human brain tissue showed that MMP-2 and -9 expression correlated with MP activation, traffic of activated macrophages, and astrogliosis in HIVE (54). In vitro assays showed that immune activation potentiated MMP production early after MP differentiation.
Chemokines likely play a critical role in HIVE promoting MP infiltration into the brain. Schmidtmayerova and colleagues showed that the -chemokines, macrophage inflammatory protein (MIP)-1 and MIP-1, are up-regulated in human MPs after HIV-1 infection. Chemokine mRNAs were found in brain cells with morphological features of MPs and in astrocytes of brain tissues infected with HIV-1. Another -chemokine, macrophage chemoattractive protein (MCP)-1, was found in brains and CSF of patients with HAD. Our recent work showed that such MP activation and, to a lesser extent, HIV-1 infection and reactive astrogliosis resulted in prominent chemokine production, macrophage brain infiltration, and neurological decline (55). An in vitro BBB model, utilized to elucidate the contribution of individual chemokines for MP migration into the brain, showed that MCP-1 was the primary chemoattractant for monocytes (55). Correlative analyses performed in an in vitro BBB system, an animal model of HIVE, and pathological analyses of postmortem brain tissue showed that microglia and astrocytes are principle sources of -chemokines controlling MP migration through the BBB in HAD (55).
MP brain infiltration also results in significant structural and functional abnormalities of microvasculature in HIV-1 CNS infection (56, 57). While Dalastra et al. demonstrated significant disruption of tight junctions of BMVEC in HIVE patients (58), we showed impairments of both tight junctions and specialized transport systems (P-glycoprotein) protecting the brain from toxic insults in association with the intensity of HIVE (59).
Acquired immunity and HAD
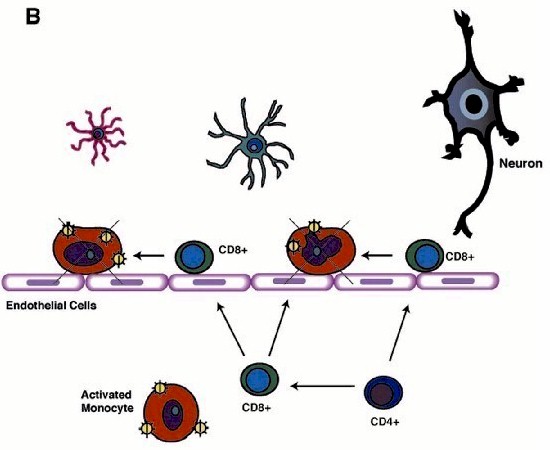
Although HIV-1 enters the brain early following viral infection, productive viral replication and brain MP invasion occurs years later and only in some infected people (2). The onset of neurological disease coincides with immunodeficiency and progression to AIDS, suggesting that virus-specific immune responses (including cytotoxic CD8+ T cells) play a protective role in the CNS (60). Defective chemotactic responses in the brain could lead to a breakdown in such protective mechanisms. Brain tissue affected by HIVE features activation and productive infection of MPs and limited lymphocyte infiltration. Since -chemokines (including IFN- -inducible 10 kDa protein [IP-10]) are responsible for attraction of T lymphocytes (expressing CXCR3 receptor), we hypothesized that -chemokine deficit could alter T cell responses in HIVE. Our in vitro experiments proved that lymphocyte CXCR3-mediated chemotactic responses remained operative (6). In human brain tissue with HIVE, we found a positive correlation between HIV-1 pol and IP-10/CD14 (MP marker)/CXCR3 (lymphocyte marker), and between CD14 and CXCR3 mRNA expression reflecting association between virus replication in MPs and MP-mediated attraction of T cells (61). CD8+ lymphocytes were found in increased numbers in HIVE brains as compared to brain tissue from seropositive patients without evidence of encephalitis. One possible explanation could be that circulating HIV-specific CD8+ T cells may be partially anergic and may be unable to eliminate HIV-1 infected cells in vivo in the setting of lacking or functionally impaired helper CD4+ T cells (62, 63). There is ample evidence to support this idea. After initial entry of infected MPs (perivascular MP infiltrates seen in humans and simian immunodeficiency virus-infected macaques), productively infected cells disappear from CNS (Fig. 1A) (64-66). Perivascular mononuclear infiltrates (mainly composed of CD8+T cells) are detected in the brain during so-called "latent" infection probably reflecting continuous egress of infected MPs and their elimination by virus-specific CD8+ T cells (Fig. 1B). Decrease of CD4+ lymphocytes in peripheral blood results in inefficient CD8+ T cell responses and massive influx of MPs driven by -chemokine production from activated microglia and astrocytes (Fig. 1C). The importance of such interactions in HAD pathogenesis is currently under investigation in our laboratories.
-inducible 10 kDa protein [IP-10]) are responsible for attraction of T lymphocytes (expressing CXCR3 receptor), we hypothesized that -chemokine deficit could alter T cell responses in HIVE. Our in vitro experiments proved that lymphocyte CXCR3-mediated chemotactic responses remained operative (6). In human brain tissue with HIVE, we found a positive correlation between HIV-1 pol and IP-10/CD14 (MP marker)/CXCR3 (lymphocyte marker), and between CD14 and CXCR3 mRNA expression reflecting association between virus replication in MPs and MP-mediated attraction of T cells (61). CD8+ lymphocytes were found in increased numbers in HIVE brains as compared to brain tissue from seropositive patients without evidence of encephalitis. One possible explanation could be that circulating HIV-specific CD8+ T cells may be partially anergic and may be unable to eliminate HIV-1 infected cells in vivo in the setting of lacking or functionally impaired helper CD4+ T cells (62, 63). There is ample evidence to support this idea. After initial entry of infected MPs (perivascular MP infiltrates seen in humans and simian immunodeficiency virus-infected macaques), productively infected cells disappear from CNS (Fig. 1A) (64-66). Perivascular mononuclear infiltrates (mainly composed of CD8+T cells) are detected in the brain during so-called "latent" infection probably reflecting continuous egress of infected MPs and their elimination by virus-specific CD8+ T cells (Fig. 1B). Decrease of CD4+ lymphocytes in peripheral blood results in inefficient CD8+ T cell responses and massive influx of MPs driven by -chemokine production from activated microglia and astrocytes (Fig. 1C). The importance of such interactions in HAD pathogenesis is currently under investigation in our laboratories.
| A | B | C |
 |
 |
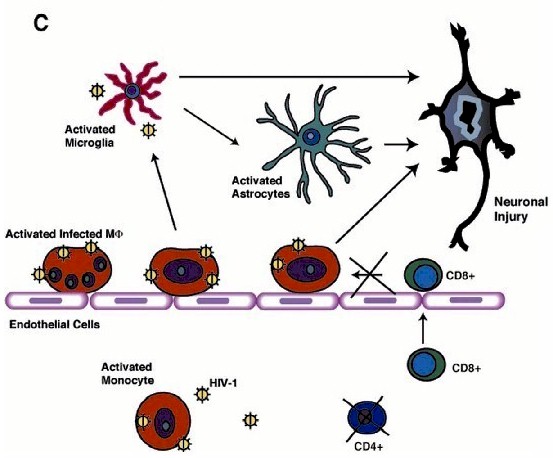 |
| Figure 1. Cell-associated traffic of HIV-1 in brain. A schematic representation of postulated events initiated by viral entry into the CNS leading to MP activation and neuronal injury are shown. (A) During period of initial viremia, HIV-1 enters brain perivascular spaces in productively infected monocytes. (B) Later, during so-called "latent" infection HIV-1-infected MP disappear from CNS due to elimination by virus specificCD8+ T cells through their efficient cooperation with CD4+ lymphocytes. (C) During profound immunosuppression, decrease of CD4+ lymphocytes in peripheral blood results in inadequate CD8+ T cell responses and massive influx of MP driven by -chemokine production from activated microglia and astrocytes resulting in HIVE and HAD. |
||
Conclusions
Issues of HIV-1 trafficking into the CNS are closely linked with entry of cells from the periphery into the CNS, their immune status and the resultant immune activation in the neural cells, molecular evolution of HIV-1 in different tissue compartments, and the changes of acquired immune responses to the virus. Although the methods of HIV-1 penetration of the CNS are diverse, there is general consensus that monocyte-associated virus is the predominant route of HIV-1 traffic in the brain. MPs deliver the virus to the brain acting as a "Trojan horse." HIV-1 enters brain perivascular spaces in productively infected monocytes during period of initial viremia. During so-called "latent" infection, HIV-1-infected MPs disappear from CNS due to elimination by virus-specific CD8+ T cells through their efficient cooperation with CD4+ lymphocytes. Decrease of CD4+ lymphocytes in peripheral blood during profound immunosuppression results in inadequate CD8+ cell responses and massive influx of MPs driven by -chemokine production from activated MPs and astrocytes. Indirect mechanisms initiated by activated MPs play a major role in neuronal dysfunction and death resulting in HAD. Neuro-immune events in HAD mainly revolve around MP secretory products (cellular and viral toxins) produced during HIVE.
References
| 1. | J. C. McArthur et al., Neurology 43, 2245 (1993). PubMed. |
| 2. | H. E. Gendelman et al., AIDS 11 (Suppl. A), S35 (1997). |
| 3. | M. Maschke et al., J. Neurol. Neurosurg. Psychiatry 69, 376 (2000). PubMed. |
| 4. | N. Sacktor et al., Neurology 56, 257 (2001). PubMed. |
| 5. | G. J. Dore et al., AIDS 13, 1249 (1999). PubMed. |
| 6. | S. Koenig et al., Science 233, 1089 (1986). PubMed. |
| 7. | G. J. Nuovo, M. L. Alfieri, Mol. Med. 2, 358 (1996). PubMed. |
| 8. | Y. Saito et al., Neurology 44, 474 (1994). PubMed. |
| 9. | E. Asare et al., Am. J. Pathol. 148, 31 (1996). PubMed. |
| 10. | E. Masliah et al., Ann. Neurol. 42, 963 (1997). PubMed. |
| 11. | I. P. Everall et al., Brain Pathol. 9, 209 (1999). PubMed. |
| 12. | J. D. Glass, H. Fedor, S. L. Wesselingh, J. C. McArthur, Ann. Neurol. 38, 755 (1995). PubMed. |
| 13. | H. S. Nottet et al., J. Immunol. 154, 3567 (1995). PubMed. |
| 14. | H. A. Gelbard et al., J. Virol. 68, 4628 (1994). PubMed. |
| 15. | M. P. Heyes et al., Brain 124, 1033 (2001). PubMed. |
| 16. | D. Giulian et al., J. Neurosci. 16, 3139 (1996). PubMed. |
| 17. | D. C. Adamson et al., Science 274, 1917 (1996). PubMed. |
| 18. | D. E. Brenneman et al., Nature 335, 639 (1988). PubMed. |
| 19. | D. R. New, M. Ma, L. G. Epstein, A. Nath, H. A. Gelbard, J. Neurovirol. 3, 168 (1997). PubMed. |
| 20. | J. Hesselgesser et al., Curr. Biol. 8, 595 (1998). PubMed. |
| 21. | J. Zheng et al., J. Neuroimmunol. 98, 185 (1999). PubMed. |
| 22. | H. Adle-Biassette et al., Neuropathol. Appl. Neurobiol. 25, 123 (1999). PubMed. |
| 23. | B. Giometto et al., Ann. Neurol. 42, 34 (1997). PubMed. |
| 24. | R. Johnson, Viral infections of the nervous system (Lippincott-Raven, Philadelphia, 1998). |
| 25. | P. Shapshak et al., AIDS Res. Hum. Retroviruses 15, 811 (1999). PubMed. |
| 26. | L. G. Epstein et al., Virology 180, 583 (1991). PubMed. |
| 27. | E. S. Hughes, J. E. Bell, P. Simmonds, J. Gen. Virol. 78, 2871 (1997). PubMed. |
| 28. | E. S. Hughes, J. E. Bell, P. Simmonds, J. Virol. 71, 1272 (1997). PubMed. |
| 29. | M. Mayne, A. C. Bratanich, P. Chen, F. Rana, A. Nath, C. Power, Neuroimmunomodulation 5, 184 (1998). PubMed. |
| 30. | A. Morris et al., J. Virol. 73, 8720 (1999). PubMed. |
| 31. | J. K. Wong et al., J. Virol. 71, 2059 (1997). PubMed. |
| 32. | B. T. Korber et al., J. Virol. 68, 7467 (1994). PubMed. |
| 33. | C. Power et al., J. Virol. 68, 4643 (1994). PubMed. |
| 34. | C. Power et al., Curr. Top. Microbiol. Immunol. 202, 89 (1995). PubMed. |
| 35. | S. M. Wolinsky et al., Science 272, 537 (1996). PubMed. |
| 36. | K. Zhang et al., Virology 283, 19 (2001). PubMed. |
| 37. | L. H. Ping et al., J. Virol. 73, 6271 (1999). PubMed. |
| 38. | A. Ghorpade et al., J. Virol. 72, 3340 (1998). PubMed. |
| 39. | A. Ghorpade et al., J. Virol. 72, 3351 (1998). PubMed. |
| 40. | J. M. Strizki et al., J. Virol. 70, 7654 (1996). PubMed. |
| 41. | D. Gabuzda, J. Wang, J. Neurovirol. 6 (Suppl. 1), S24 (2000). PubMed. |
| 42. | D. Gabuzda, J. Wang, J. Neurovirol. 5, 643 (1999). PubMed. |
| 43. | Y. Huang et al., Nat. Med. 2, 1240 (1996). PubMed. |
| 44. | W. A. Paxton et al., Nat. Med. 2, 412 (1996). PubMed. |
| 45. | T. K. Smit et al., Virology 279, 509 (2001). PubMed. |
| 46. | J. Martin, C. C. LaBranche, F. Gonzalez-Scarano, J. Virol. 75, 3568 (2001). PubMed. |
| 47. | S. Li et al., J. Virol. 73, 9741 (1999). PubMed. |
| 48. | H. E. Gendelman et al., J. Infect. Dis. 178, 1000 (1998). PubMed. |
| 49. | G. Poli, Eur. J. Clin. Invest. 29, 723 (1999). PubMed. |
| 50. | H. S. Nottet et al., J. Immunol. 156, 1284 (1996). PubMed. |
| 51. | L. Pulliam, R. Gascon, M. Stubblebine, D. McGuire, and M. S. McGrath, Lancet 349, 692 (1997). PubMed. |
| 52. | Y. Persidsky, M. Buttini, J. Limoges, P. Bock, H. E. Gendelman, J. Neurovirol. 3, 401 (1997). PubMed. |
| 53. | B. Sporer et al., J. Infect. Dis. 178, 854 (1998). PubMed. |
| 54. | A. Ghorpade et al., J. Virol. 75, 6572 (2001). PubMed. |
| 55. | Y. Persidsky et al., Am. J. Pathol. 155, 1599 (1999). PubMed. |
| 56. | C. K. Petito, K. S. Cash, Ann. Neurol. 32, 658 (1992). PubMed. |
| 57. | A. Buttner, P. Mehraein, S. Weis, Acta Neuropathol. (Berl) 92, 35 (1996). PubMed. |
| 58. | L. M. Dallasta et al., Am. J. Pathol. 155, 1915 (1999). PubMed. |
| 59. | Y. Persidsky, J. Zheng, D. Miller, H. E. Gendelman, J. Leukoc. Biol. 68, 413 (2000). PubMed. |
| 60. | S. Sopper et al., J. Virol. 72, 9940 (1998). PubMed. |
| 61. | L. Poluektova et al., J. Neuroimmunol. 120, 112 (2001). PubMed. |
| 62. | C. J. Pitcher et al., Nat. Med. 5, 518 (1999). PubMed. |
| 63. | L. A. Trimble, P. Shankar, M. Patterson, J. P. Daily, J. Lieberman, J. Virol. 74, 7320 (2000). PubMed. |
| 64. | L. E. Davis et al., Neurology 42, 1736 (1992). PubMed. |
| 65. | E. Sinclair, F. Gray, A. Ciardi, F. Scaravilli, J. Neuropathol. Exp. Neurol. 53, 43 (1994). PubMed. |
| 66. | L. Chakrabarti et al., Am. J. Pathol. 139, 1273 (1991). PubMed. |
| 67. | We thank Ms. Robin Taylor and Ms. Radika Suryadevara for excellent editorial support. Supported in part by NIH grants AI42404-04 R29 (Y.P.). |
| Copyright Information | Site map |
